Pocket Medicine: The Massachusetts General Hospital Handbook of Internal Medicine (63 page)
Read Pocket Medicine: The Massachusetts General Hospital Handbook of Internal Medicine Online
Authors: Marc Sabatine
Tags: #Medical, #Internal Medicine

• Treatment: treat underlying disease ± iron and/or erythropoiesis-stimulating agent (ESA, eg, Epo). Iron if ferritin <100 or Fe/TIBC <20%. Consider ESA if Epo <500. Avoid ESA in cancer if treatment goal is cure (
Leuk Res
2012;36:939). Unclear if one should treat highly sx Pts w/ goal Hb 10–12 g/dL; weigh risk of thrombosis.
Anemias of chronic disorders
• Anemia of chronic inflammation (see above) • Anemia of chronic kidney disease: ↓ Epo; treat w/ Epo (see “Chronic Kidney Disease”) • Endocrine deficiencies: hypometabolism and ↓ O
2
demand with thyroid, pituitary, adrenal, or parathyroid disease → ↓ Epo; can be normocytic or macrocytic
Sideroblastic anemia
(see above)
Pure red cell aplasia
• Destructive antibodies or lymphocytes → ineffective erythropoiesis • Associated with thymoma, CLL and parvovirus infection • Diagnostic studies:
lack of erythroid precursors on BM bx
, other lines normal • Treatment: thymectomy if thymus enlarged; IVIg if parvovirus infection; immunosuppression if CLL or idiopathic; supportive care with PRBC transfusions; ? erythropoietin receptor agonist if due to antierythropoietin Ab (
NEJM
2009;361:1848)
MACROCYTIC ANEMIAS
includes megaloblastic and nonmegaloblastic causes
Megaloblastic anemia
•
Impaired DNA synthesis
→ cytoplasm matures faster than nucleus → ineffective erythropoiesis and macrocytosis; due to
folate
or
B12 deficiency
;
MDS
• ✓
folate
and
vitamin B12
; ↑ LDH & indirect bilirubin (due to ineffective erythropoiesis) • Smear:
neutrophil hypersegmentation
,
macro-ovalocytes
, anisocytosis, poikilocytosis
Folate deficiency
• Folate present in leafy green vegetables and fruit; total body stores sufficient for
2–3 mo
• Etiologies:
malnutrition
(alcoholics, anorectics, elderly), ↓ absorption (sprue), impaired metabolism (methotrexate, pyrimethamine, trimethoprim), ↑ requirement (chronic hemolytic anemia, pregnancy, malignancy, dialysis) • Diagnosis: ↓ folate; ↓ RBC folate, ↑ homocyst. but nl methylmalonic acid (unlike B
12
defic.) • Treatment: folate 1–5 mg PO qd for 1–4 mo or until complete hematologic recovery;
critical to r
/
o B
1
2
deficiency first (see below)
Vitamin B
12
deficiency
(
NEJM
2013;368:149)
• B
12
present only in foods of animal origin; total body stores sufficient for
2–3 y
• Binds to
intrinsic factor
(IF) secreted by gastric parietal cells; absorbed in terminal ileum • Etiologies: malnutrition (alcoholics, vegans),
pernicious anemia
(PA, autoimmune disease against gastric parietal cells, a/w polyglandular endocrine insufficiency and ↑ risk of gastric carcinoma), other causes of ↓ absorption (gastrectomy, sprue, Crohn’s disease), ↑ competition (intestinal bacterial overgrowth, fish tapeworm) • Clinical manifestations:
neurologic
changes (
subacute combined degeneration
) affecting peripheral nerves, posterior and lateral columns of the spinal cord and cortex → numbness, paresthesias, ↓ vibratory and positional sense, ataxia, dementia • Dx: ↓ B
12
; ↑ homocysteine and methylmalonic acid; anti-IF Ab; Schilling test; ↑ gastrin in PA • Treatment: 1 mg B
12
IM qd × 7 d → q wk × 4–8 wk → q month for life neurologic abnormalities are reversible if treated w/in 6 mo folate can reverse
hematologic
abnormalities of B
12
deficiency but not
neurologic
changes (and can lead to “steal” of B
12
stores → worsening of neuro complications) oral supplementation (2 mg qd) appears feasible as well (
Blood
1998;92:1191) even w/o IF
Nonmegaloblastic macrocytic anemias
•
Liver disease
: often macrocytic, may see target cells •
Alcoholism
: BM suppression & macrocytosis independent of folate/B
12
defic. or cirrhosis •
Reticulocytosis
•
Other causes
: hypothyroidism; MDS; meds that impair DNA synthesis (zidovudine, 5-FU, hydroxyurea, Ara-C); hereditary orotic aciduria; Lesch-Nyhan syndrome.
PANCYTOPENIA
Etiologies
• Hypocellular bone marrow (nl cellularity ~100 – age):
aplastic anemia
, hypoplastic MDS
• Cellular bone marrow:
MDS
, aleukemic leukemia, PNH, severe megaloblastic anemia • Marrow replacement (myelophthisis):
myelofibrosis
, metastatic solid tumors, granulomas • Systemic diseases: hypersplenism, sepsis, alcohol, toxins
Clinical manifestations
• Anemia → fatigue • Neutropenia → recurrent infections • Thrombocytopenia → mucosal bleeding & easy bruisability
Aplastic anemia
= stem cell failure (
Lancet
2005;365:1647;
Blood
2012;120:1185)
• Epidemiology: 2–5 cases/10
6
/y; biphasic (major peak in adolescents, 2nd peak in elderly) • Diagnosis: pancytopenia w/ ↓ retics, BM bx w/ cytogenetics showing hypocellularity • Etiologies:
idiopathic
(
1
/
2
–
1
/
3
of cases)
stem cell destruction
:
radiation
,
chemotherapy
,
chemicals
(eg, benzene) idiosyncratic
med rxn
(eg, chloramphenicol, NSAIDs, sulfa drugs, gold, carbamazepine, antithyroid)
viruses
(HHV-6, HIV, EBV, parvovirus B19); also
post-hepatitis
(non A, B or C)
immune disorders
(SLE, GVHD post-HSCT, thymoma)
PNH (see below); Fanconi’s anemia (congenital disorder w/ pancytopenia, macrocytic anemia, ↑ risk of MDS, AML, & SCC of head & neck, and multiple physical anomalies);
shortened telomeres: seen w/ telomerase (
TERT, TERC
) mut (10% of aplastic anemia), dyskeratosis congenita/DKC1 mut; a/w IPF, cirrhosis (
NEJM
2009;361:2353)
• Treatment and prognosis
allogeneic HSCT
: for
young
Pts → ~80% long-term survival and significantly ↓ risk of malignant evolution, but has risk of transplant-related morbidity & mortality; if possible avoid transfusions (and alloimmunization) pretransplant
immunosuppression
(CsA/tacrolimus, ATG): 70–80% respond, with 80–90% 5-y survival in responders (96% vs. 76% w/ horse vs. rabbit ATG;
NEJM
2011;365:430); 15–20% 10-y incidence of clonal disorders (mostly MDS, AML, PNH)
TPO mimetics
(eg, eltrombopag) may be option in refractory disease (
NEJM
2012;367:11)
supportive care:
transfusions, antibiotics, possible utility of G-CSF and Epo
Myelodysplastic syndromes (MDS)
(qv)
Paroxysmal nocturnal hemoglobinuria (PNH)
(
Blood
2009;113:6522)
• Acquired clonal stem cell disorder = inactivating somatic mutation of
PIG-A
gene → deficiency of GPI-anchor for CD55 & CD59 (inhib of complement) → complement-mediated RBC lysis, plt aggreg., & hypercoagulability • Clinical: intravascular
hemolytic anemia
,
hypercoagulability
(venous > arterial; esp. intraabdominal, cerebral), smooth muscle dystonias,
deficient hematopoiesis
(cytopenias); a/w aplastic anemia, MDS and evolution to AML
• Dx:
flow cytometry
(↓ CD55 & CD59) on RBCs and granulocytes; urine hemosiderosis • Treatment: supportive care (iron, folate, transfusions); consider anti-coagulation allogeneic HSCT for hypoplasia or severe thrombosis eculizumab (Ab inactivates terminal complement C5s): ↓ hemolysis, improves QoL & stabilizes Hb levels (
NEJM
2004;350:552 & 2006;355:1233;
Lancet
2009;373:759); must have meningococcal vaccination
Myelophthisic anemia
(see also “Primary Myelofibrosis”)
• Infiltration of bone marrow by cancer, leukemia, infection, fibrosis (primary myelofibrosis), granulomas, lysosomal storage disorders
HEMOLYTIC ANEMIAS
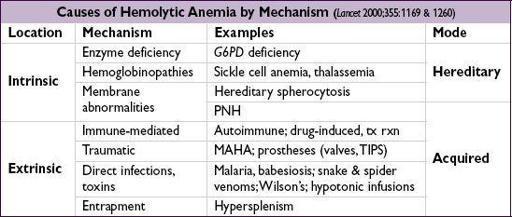
Diagnostic evaluation
• ↑ reticulocyte count (RI >2%), ↑ LDH, ↓ haptoglobin (83% Se, 96% Sp), ↑ indirect bili • Autoimmune hemolysis: Coombs’ test = direct antiglobulin test (DAT) →if agglutination occurs when antisera against Ig or C3 are applied to patient RBCs • Intravascular: ↑↑ LDH, ↓↓ haptoglobin; hemoglobinemia, hemoglobinuria, hemosiderinuria • Extravascular: splenomegaly • Family h/o anemia; personal or family h/o cholelithiasis
Glucose-6-phosphate dehydrogenase (G6PD) deficiency
(
Lancet
2008;371:64)
• X-linked defect of metabolism (
G6PD
mutations) w/ ↑ susceptibility to oxidative damage • Most common inof African or Mediterranean descent (malaria-endemic areas) • Hemolysis precipitated by
drugs
(sulfonamides, dapsone, primaquine, doxorubicin, methylene blue),
infection
,
DKA
or
foods
(fava beans in children) • Diagnosis: smear may show RBC
Heinz bodies
(oxidized Hb) that result in
bite cells
once removed by spleen; ↓ G6PD levels (
may be normal after acute hemolysis
as older RBCs have already lysed and young RBCs may still have near normal levels)
Sickle cell anemia
(
Lancet
2010;376:2018)
• Recessive β-globin mutation → structurally abnl hemoglobin (HbS). ~8% African Americans heterozygotes (“sickle trait”; usually w/o sx); ~1/400 homozygotes (sickle cell disease).