Pocket Medicine: The Massachusetts General Hospital Handbook of Internal Medicine (62 page)
Read Pocket Medicine: The Massachusetts General Hospital Handbook of Internal Medicine Online
Authors: Marc Sabatine
Tags: #Medical, #Internal Medicine

r/o UTI and non-GU causes (GI or vaginal bleed)
Urine cytology (Se
70%, Sp
95%), not adequate substitute for cystoscopy
Renal imaging: helical CT ± contrast (r/o nephrolithiasis and neoplasia of upper tract), cystoscopy (r/o bladder neoplasia, esp. ≥35 y), ± MRI, retrograde pyelogram, U/S
NEPHROLITHIASIS
Types of stones and risk factors
(
J Clin Endocrinol Metabol
2012;97:1847)
•
Calcium
(Ca oxalate > Ca phosphate):
70–90% of kidney stones
Urine findings: ↑ Ca, ↑ oxalate (Ca-ox only), ↑ pH (Ca-phos only), ↓ citrate, ↓ volume
2° hypercalciuria: 1° hyperparathyroidism, distal RTA, sarcoid
2° hyperoxaluria: Crohn’s, ileal disease w/ intact colon, gastric bypass
Diet: ↑ animal protein, ↑ sucrose, ↑ Na, ↓ K, ↓ fluid, ↓ fruits/vegetables, ↑ vit. C, ↓ Ca
•
Uric acid
: 5–10% of kidney stones, radiolucent on plain film
Urine findings: ↑ uric acid, ↓ pH (eg, from chronic diarrhea)
•
Magnesium ammonium phosphate
(“struvite” or “triple phosphate”)
Chronic upper UTI w/ urea-splitting organisms (eg,
Proteus, Klebs
) → ↑ urine NH
3
, pH >7
•
Cystine
: inherited defects of tubular amino acid reabsorption
Clinical manifestations
• Hematuria (absence does not exclude diagnosis), flank pain, N/V, dysuria, frequency • Ureteral obstruction (stones >5 mm unlikely to pass spont.) → AKI if solitary kidney • UTI: ↑ risk of infection proximal to stone; urinalysis of distal urine may be normal
Workup
•
Noncontrast helical CT scan
(ureteral dilation w/o stone suggests recent passage) 97% sens. 96% spec. (
AJR
2008;191:396) • Strain urine for stone to analyze; U/A & UCx; electrolytes, BUN/Cr, Ca, PO
4
, PTH
• 24-h urine × 2 (>6 wk after acute setting) for Ca, PO
4
, oxalate, citrate, Na, Cr, pH, K, vol.
Acute treatment
(
NEJM
2004;350:684)
• Analgesia (narcotics ± NSAIDs; combination superior,
Ann Emerg Med
2006;48:173), ensure adequate fluid repletion, antibiotics if UTI • Consider alpha blocker > CCB to promote ureteral relaxation (
Lancet
2006;368:1171) • Indications for
immediate urologic eval and
/
or hosp
: obstruction (esp. solitary or transplant kidney), urosepsis, intractable pain or vomiting, significant AKI • Urologic Rx: lithotripsy (
NEJM
2012:367:50), stent, perc nephrostomy, ureteroscopic removal
Chronic treatment
(
J Clin Endocrinol Metabol
2012;97:1847)
• Increase fluid intake (>2 L/d) for goal UOP 2 L/d • Calcium stones: 24-h urine identifies
specific urinary risk factors to treat
↓ Na and meat intake (
NEJM
2002;346:77), thiazides: decrease urine Ca
Depending on 24-h urine: K-citrate, dietary oxalate restriction, allopurinol
High dietary Ca is likely beneficial by ↓ oxalate absorp., unclear role of Ca supplements
• Uric acid: urine alkalinization (K-citrate), allopurinol • Magnesium ammonium phosphate: antibiotics to treat UTI, urologic intervention, acetohydroxamic acid: urease inhibitor, reserve for experienced clinician, poorly tolerated • Cystine: urine alkalinization (K-citrate), D-penicillamine, tiopronin
NOTES
ANEMIA
↓
in RBC mass: Hct
<
41% or Hb
<
13.5 g/dL (men); Hct
<
36% or Hb
<
12 g/dL (women)
Clinical manifestations
• Symptoms: ↓ O
2
delivery → fatigue, exertional dyspnea, angina (if CAD) • Signs: pallor (mucous membranes, palmar creases), tachycardia, orthostatic hypotension • Other findings:
jaundice
(hemolysis),
splenomegaly
(thalassemia, neoplasm, chronic hemolysis),
petechiae
/
purpura
(bleeding disorder),
glossitis
(iron, folate, vitamin B
12
defic.),
koilonychia
(iron defic.),
neurologic abnormalities
(B
12
defic.)
Diagnostic evaluation
• History: bleeding, systemic illness, drugs, exposures, alcohol, diet (including
pica
), FHx • CBC w/ diff.; RBC params incl. retics, MCV (nb, mixed disorder can → nl MCV), RDW
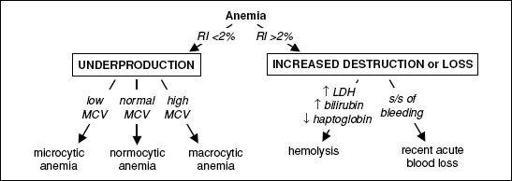
•
Reticulocyte index
(RI) = [reticulocyte count × (Pt’s Hct/nl Hct)]/maturation factor maturation factors for a given Hct: 45% = 1, 35% = 1.5, 25% = 2, 20% = 2.5
RI >2% → adequate marrow response; RI <2% → hypoproliferation •
Peripheral smear
: select area where RBCs evenly spaced and very few touch each other; ✓ RBC size, shape, inclusions (see Appendix & Peripheral Smear inserts), WBC morphology, plt count • Additional labs as indicated: hemolysis labs (if RI >2%), iron/TIBC, ferritin, folate, B
12
, LFTs, BUN and Cr, TFTs, Hb electrophoresis, enzyme analyses, gene mutation screens •
Bone marrow (BM) aspirate and biopsy (bx)
with cytogenetics as indicated
Figure 5-1 Approach to anemia
MICROCYTIC ANEMIAS
Figure 5-2 Approach to microcytic anemias
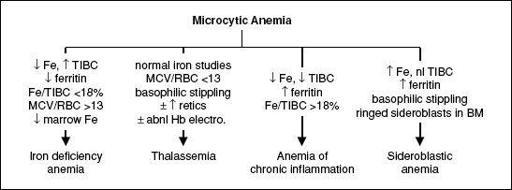
Iron deficiency
(
NEJM
1999;341:1986;
Gut
2011;60:1309)
• ↓ marrow iron & depleted body iron stores → ↓ heme synthesis → microcytosis → anemia • Special clinical manifestations: angular cheilosis, atrophic glossitis, pica (consumption of nonnutritive substances such as ice, clay), koilonychia (nail spooning) Plummer-Vinson syndrome (iron deficiency anemia, esophageal web & atrophic glossitis) • Etiologies:
chronic bleeding
(GI—incl. cancer, menstrual, parasites, etc.), ↓
supply
(malnutrition; ↓ absorp. due to celiac sprue, Crohn’s, ↑ gastric pH, subtotal gastrectomy), ↑
demand
(preg., Epo). Rare Fe refractory genetic disorder due to hepcidin dysregulation (
Nat Genet
2008;40:569).
• Diagnosis: ↓
Fe
, ↑
TIBC
, ↓
ferritin
(esp. <15), ↓
transferrin sat
(Fe/TIBC; esp. <15%), ↑ soluble transferrin receptor; ↑ plt; unless hx c/w other etiology,
initiate workup for GIB
; incl.
H. pylori
serology, ? celiac sprue labs (
anti-TTG
, antigliadin, antiendomysial Ab) • Treatment (Fe supplementation): oral Fe tid (~6 wk to correct anemia; ~6 mo to replete Fe stores); in cases of excessive/persistent GI losses or for dialysis or cancer Pts prior to Epo Rx, IV iron (Fe-sucrose, -gluconate, -dextran) should be considered
Thalassemias
(
Lancet
2013;379:373)
• ↓ synthesis of ɑ-or β-globin chains of Hb → ≠ subunits → destruction of RBCs and erythroid precursors; ∴ anemia from hemolysis
and
ineffective erythropoiesis •
ɑ-thalassemia
: deletions in ɑ-globin gene complex on chr. 16 (nl 4 ɑ genes)
3 ɑ → ɑ-thal-2 trait = silent carrier; 2 ɑ → ɑ-thal-1 trait or ɑ-thal minor = mild anemia
1 ɑ → HbH (β
4
) disease = severe anemia, hemolysis and splenomegaly
0 ɑ genes → Hb Barts (γ
4
) = intrauterine hypoxia and hydrops fetalis •
β-thalassemia
: mutations in β-globin gene on chr. 11 → absent or ↓ gene product
1 mutated β gene → thal minor (or trait) = mild anemia (no transfusions)
2 mutated β genes → thal intermedia (occasional transfusions) or thal major ( = Cooley’s anemia; transfusion dependent) depending on severity of mutations • Special clinical manifestations (in severe cases): chipmunk facies, pathologic fractures, hepatosplenomegaly (due to extramedullary hematopoiesis), high-output CHF, bilirubin gallstones, iron overload syndromes (from chronic transfusions) • Diagnosis: MCV <70,
normal Fe
,
MCV
/
RBC count
<
13
[Mentzer Index, 60% Se, 98% Sp; (
Ann Hem
2007;86:486)], ± ↑ retics, basophilic stippling;
Hb electrophoresis
: ↑ HbA
2
(ɑ
2
δ
2
) in β-thal;
normal
pattern in ɑ-thal trait • Treatment: folate; transfusions + deferoxamine, deferasirox (oral iron chelator); splen-ectomy if ≥50% ↑ transfusions; consider allo-HSCT in children w/ severe β-thal major
Anemia of chronic inflammation
(see below)
Sideroblastic anemia
• Defective heme biosynthesis within RBC precursors • Etiologies:
hereditary/X-linked
(
ALAS2
mutations),
idiopathic
,
MDS-RARS
,
reversible
(alcohol, lead, isoniazid, chloramphenicol, copper deficiency, hypothermia) • Special clinical manifestations: hepatosplenomegaly, iron overload syndromes • Dx: review social, work & TB hx; can be microcytic, normocytic or macrocytic; variable pop of hypochromic RBCs; ↑ Fe, nl TIBC, ↑ ferritin, basophilic stippling, RBC
Pappenheimer bodies
(Fe-containing inclusions),
ring sideroblasts
(w/ iron-laden mitochondria) in BM
• Treatment: treat reversible causes; trial of pyridoxine, supportive transfusions for severe anemia; high-dose pyridoxine for some hereditary cases
NORMOCYTIC ANEMIAS
Pancytopenia
(see below)
Anemia of chronic inflammation
(ACI;
NEJM
2005;352:1011; 2009;361:1904)
• ↓ RBC production due to impaired iron utilization and functional iron deficiency from ↑
hepcidin
; cytokines (IL-6, TNF-a) cause ↓ Epo responsiveness/production • Etiologies: autoimmune disorders, chronic infection, inflammation, HIV, malignancy • Dx: ↓
Fe
, ↓
TIBC (usually normal or low transferrin sat)
, ± ↑
ferritin
; usually normochromic, normocytic (~70% of cases) but can be microcytic if prolonged • Coexisting iron deficiency common. Dx clues include ↓ serum ferritin levels, absence of iron staining on BM bx, response to a trial of oral iron and/or ↑ soluble transferrin receptor/ferritin index (
response to a trial of oral iron and/or ↑ soluble transferrin receptor/ferritin index (
Blood
1997;89:1052).