Junk DNA: A Journey Through the Dark Matter of the Genome (40 page)
Read Junk DNA: A Journey Through the Dark Matter of the Genome Online
Authors: Nessa Carey

d
The drug is called Mipomersen, also known as Kynamro.
20. Some Light in the Darkness
As we near the end of our wanderings through the darker regions of our genome, the more alert reader may remember that we haven’t addressed the mystery of one of the human disorders first encountered at the beginning of this book. This condition is the cumbersomely named facioscapulohumeral muscular dystrophy, or FSHD. This is the condition in which there is wasting of the muscles of the face, shoulders and upper arms.
It occurs when patients inherit a smaller number of a particular genetic repeat on one of their copies of chromosome 4. Even quite a few years after the mutation was identified, the reason why this caused disease remained mysterious, because there just didn’t seem to be a protein-coding gene anywhere near the genetic defect.
We finally have an understanding of how the disease symptoms are caused and the story is remarkable. It pulls together a number of the themes we have already encountered, showing how junk DNA, epigenetics, genetic fossils and abnormal RNA processing all work together to create an extraordinary tale of pathological conspiracy.
1
Let’s recap a little. On normal copies of chromosome 4, a region is repeated between eleven and 100 times. This region is just over 3,000 base pairs in length. In people with FSHD, there is a much smaller number of repeats – between one and ten units – on one of their copies of the chromosome.
Here’s where the first complication arises. There are people who have ten or fewer copies of this unit, but who don’t have
FSHD. Their muscles are completely healthy. The low number of repeats only causes a problem if it occurs on a copy of chromosome 4 that also contains another feature.
To understand the importance of this other feature, we need to look in more detail at what is found in the repeating units. They all contain a retrogene.
a
A retrogene is a form of junk DNA. It is created when the messenger RNA from a normal cellular gene gets copied back into DNA and reinserted into the genome. It’s very similar to the process we saw in Figure 4.1 (
page 38
) and occurred long ago in human evolution.
Because retrogenes are originally created from messenger RNA templates, they often don’t include the proper regulatory sequences of normal genes. They won’t contain splicing signals (because the messenger RNA template had already been spliced before it was copied into DNA) and they lack appropriate promoter and enhancer regions. But some can still be used to produce messenger RNA. This is the case with the FSHD retrogene, but it doesn’t usually matter, because the RNA doesn’t function properly in the cell. It doesn’t contain the correct signals for adding a string of A bases to the end of the messenger RNA, the process described in Figure 16.5 (
page 233
). Because of this, the messenger RNA is unstable and doesn’t get used as a template for production of protein.
But, when a person has only a small number of FSHD repeats, and other sequences on chromosome 4 are present, the final copy of the FSHD retrogene can be spliced to an additional sequence. This creates a signal at the end of the messenger RNA which allows the cellular machinery to add A bases. This in turn stabilises the messenger RNA, and it is transported to the ribosomes to act as the template for production of a protein – a protein that should never be switched on in mature muscle cells.
The FSHD protein is one that regulates the expression of other genes by binding to specific DNA sequences. It is usually only expressed in the germline, the cells that produce eggs or sperm. There is no definitive explanation yet for why expression of this protein causes muscle wasting, and it may be that a number of mechanisms are involved. It may activate genes that trigger muscle cell death. It may cause loss of muscle stem cells, perhaps by activating other retrogenes and genomic invaders that should be kept silent. One intriguing possibility is that the muscle cells that express the FSHD protein are destroyed by the patient’s own immune system.
The germline is a tissue that is known as immunologically privileged, because normally it is kept isolated from the cells of our immune system. This means that our immune system never learns that the cells of immunologically privileged sites are a normal part of our body. If proteins from the germline are switched on in adult muscle cells, the immune system may respond as if they are foreign organisms and attack the cells that express these previously unencountered elements.
So FSHD provides us with an example of the importance of junk DNA in disease. A genetic defect changes the amount of junk DNA. As a consequence of this, a junk element is expressed and modified by the addition of a junk sequence. But there is yet more to the picture. The FSHD retrogene only becomes stably expressed in the presence of a particular pattern of epigenetic modifications.
In normal cells, the FSHD repeats are usually expressed when the cells are in a pluripotent state, such as embryonic stem cells. At this stage, the FSHD repeats are covered with activating epigenetic modifications. But as the cells differentiate, the activating modifications are replaced by repressive ones, and the region is silenced. But if pluripotent cells are created from FSHD patients, the activating modifications aren’t replaced as the cells differentiate, and the repeats remain switched on.
Another aspect of the picture is the overall control of the FSHD genetic domain. There is an insulator region between the repeated region and the rest of chromosome 4. The protein 11-FINGERS (
see page 178
) binds to this region and ensures that different patterns of epigenetic modifications are maintained in the FSHD domain compared with the adjacent areas of the chromosome.
On top of all these features, the three-dimensional structure of the relevant regions of chromosome 4 also plays a role in the expression of the FSHD retrogene. It’s almost certainly the combination of all these factors that results in the restricted pattern of muscle wasting that we see in patients with FSHD. All of these aspects have to be right (or perhaps wrong) for the symptoms to develop.
The mechanism by which a change in a junk region leads to disease in FSHD is a stunning example of the complex and multilayered ways in which the different elements of our genome work together. It also demonstrates how we need to think not in terms of linear pathways when we consider what is happening in our cells, but in terms of complex interlocking processes. Figure 20.1 demonstrates this graphically. It exemplifies why the arguments about which is the most important feature of our genome are ultimately sterile. If we disrupt any aspect there will be consequences. Some will be bigger than others, but all work together.
Of course, this doesn’t mean that every single one of our billions of base pairs has a function. Some may truly just be genomic garbage, with no utility, whereas other regions are junk in the sense that they could have been discarded but instead have been turned into something useful.
2
There is still a lot we don’t know, including some questions that we might think are very straightforward. We haven’t even got a definitive answer for how many functional regions of junk DNA exist in a cell. That might seem easy to answer but have a quick
look at Figure 20.2 and then answer the following question. How many squares are there on a chessboard?
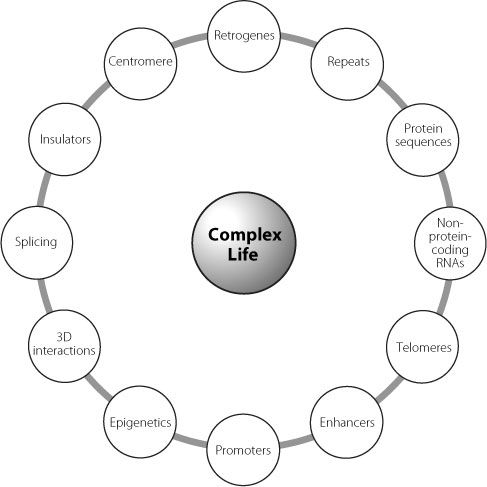
Figure 20.1
Just some of the interacting factors that have to work together to create the great organism that is you.
The instant instinctive response is always 64. But the actual answer is 204, because we can draw bigger squares of various sizes around the more obvious single black and white ones. Our genome is like that. One stretch of DNA can include a protein-coding gene, long non-coding RNAs, smallRNAs, antisense RNAs, splice signal sites, untranslated regions, promoters and enhancers. Layer on to this the effects of variations in DNA sequence between individuals, directed and random epigenetic modifications, changeable three-dimensional interactions, plus binding to other RNAs and proteins; then add in the effects of our constantly altering environment.

Figure 20.2
Quickly now, how many squares are on a chessboard?
When we really think about the complexity of our genomes, it isn’t surprising that we can’t understand everything yet. The astonishing triumph is that we understand any of it. There is always something new to be learnt, out there in the dark.
Footnote
a
This particular retrogene is called DUX4.
Notes
Chapter 1
1
. For information on the disorder and its genetics see
www.omim.org
record #160900
2
. For more information see
http://ghr.nlm.nih.gov/condition/myotonic-dystrophy
3
. For more information see
http://www.ninds.nih.gov/disorders/friedreichs_ataxia/detail_friedreichs_ataxia.htm
4
. For more information see
http://ghr.nlm.nih.gov/condition/facioscapulohumeral-muscular-dystrophy
Chapter 2
1
.
http://www.escapistmagazine.com/news/view/113307-Virtual-Typewriter-Monkeys-Pen-Complete-Works-of-Shakespeare-Almost
2
. Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion.
Science
. 1996 Mar 8;271(5254):1423–7
3
. Bidichandani SI, Ashizawa T, Patel PI. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure.
Am J Hum Genet
. 1998 Jan;62(1):111–21
4
. Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin.
Science
. 1997 Jun 13;276(5319):1709–12
5
. Kremer EJ, Pritchard M, Lynch M, Yu S, Holman K, Baker E, Warren ST, Schlessinger D, Sutherland GR, Richards RI. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n.
Science
. 1991 Jun 21;252(5013):1711–4
6
. Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome.
Cell
. 1991 May 31;65(5):905–14
7
. Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL. Absence of expression of the FMR-1 gene in fragile X syndrome.
Cell
. 1991 Aug 23;66(4):817–22