Junk DNA: A Journey Through the Dark Matter of the Genome (35 page)
Read Junk DNA: A Journey Through the Dark Matter of the Genome Online
Authors: Nessa Carey

Affected babies seem perfectly healthy at first but within a year their growth rate slows dramatically, and they remain underweight and short for the rest of their lives. The children begin developing many symptoms of old age, including thinning hair, stiffness and baldness. Although there are some ageing conditions that they don’t develop, such as Alzheimer’s disease (and the children also don’t have learning disabilities), the affected individuals do develop severe cardiovascular disease. This is usually what causes death by the early teens, as a consequence of heart attacks or major strokes.
In 2003 researchers identified the gene mutation that causes Hutchinson-Gilford Progeria. Every patient they tested had a de novo mutation, meaning one that developed spontaneously in the parents’ egg or sperm. Incredibly, in eighteen unrelated patients (out of twenty who were assessed) the mutation was exactly the same.
16
A sequence that should read GGC in a particular gene had mutated and now read as GGT. This mutation was in the amino acid-coding part of the gene. This might seem like a straightforward case of a mutation changing an amino acid in a protein, so of course the first thing to do is to look at the genetic code and see what these two sequences code for. GGC, the normal sequence, codes for a simple amino acid called glycine. But the mutated sequence, GGT, codes for – wait for it – glycine. Yep, same amino acid.
This is because our genetic code has a level of redundancy. Our genome is composed of four letters – A, C, G and T (or U in RNA). Blocks of three letters are used to code for an amino acid. There are 64 possible combinations of three from four letters. Three of these combinations are stop signals, telling the ribosomes not to add any more amino acids to a protein chain. This leaves 61 combinations to code for amino acids. But our proteins only contain 20 different amino acids. So some amino acids can be coded for by different three-letter combinations. At one extreme,
glycine is coded for by GGA, GGC, GGG and GGT(U). At the other, the amino acid methionine is only coded for by the combination AT(U)G.
But if the amino acid sequence encoded by the mutated gene doesn’t change in Hutchinson-Gilford Progeria, what causes the dramatic phenotype in this condition? Look again at Figure 17.5. The two-base sequence at the beginning of each intervening junk region within a gene is GT. In the patients where the normal GGC changes to GGT, the amino acid region gains an inappropriate extra splice signal. In the context of all the other splicing signals in that genomic region, this inappropriately positioned GT acts strongly. The spliceosome cuts the messenger RNA in the amino acid-coding region rather than in the junk region. The amino acid-coding regions join up badly and the end result is a loss of about 50 amino acids from the end of the protein. This in turn means that the protein itself isn’t processed properly, and it begins to wreak havoc in the cells. We still don’t know exactly how this leads to the extraordinary ageing we see in these children, but our best guess at the moment is that the cell nucleus isn’t maintained properly. This may lead to changes in gene expression and nuclear breakdown. Some genes and some cell types may be more sensitive to this than others.
There is another condition that affects young children called Spinal Muscular Atrophy. In this condition, the nerve cells supplying the muscles gradually die off, leading to muscle wasting and loss of mobility. There are a number of different forms, and in the most severe version the life expectancy for affected babies is very low, less than eighteen months.
17
It is relatively common for a genetic disease: in the UK about one in 40 people is a carrier, meaning about 1.5 million of us have one defective copy of the gene. Luckily, both copies of the gene have to be mutated for symptoms to develop.
18
Spinal Muscular Atrophy is caused by the deletion or loss of
function of a gene called SMN1. If we look at the human genome we might be surprised that this has such a major effect, because there is another gene that codes for exactly the same protein. This gene is called SMN2. This raises a really obvious question: since they code for the same protein, why can’t the SMN2 gene compensate for a damaged/deleted SMN1 gene?
In a rather similar way to Hutchinson-Gilford Progeria, the SMN2 has one subtle variation from SMN1. This is a change in DNA sequence in an amino acid-coding region. It doesn’t change the amino acid sequence, because it is in one of the three-base sequences with redundancy in terms of amino acid coding. Instead it changes one of the sites that help ribosomes to work out where to splice the messenger RNA molecule.
19
It doesn’t change a splice site, it changes one of the sites that influences where splicing occurs. This results in skipping of an amino acid-coding region, and the production of a protein that isn’t functional. Because of this, the SMN2 gene can’t compensate for the malfunctioning SMN1 gene. The normal SMN1 protein is required for spliceosome activity. So essentially, a mutation in one gene leads to problems with overall splicing of messenger RNAs, and this could be overcome except for an independent splicing issue with a potentially compensatory gene.
Manipulating splicing for therapeutic gain
As we saw in Chapter 7, in Duchenne muscular dystrophy, the severe muscle wasting disease carried on the X chromosome, the dystrophin gene is mutated (
see page 92
). This gene is exceptionally large, stretching for almost 2.5 million base pairs. It contains nearly 80 amino acid-coding regions, which need to be spliced and processed properly. This is particularly important because the dystrophin protein is long-lasting. This means that any change that increases the chance of mis-splicing will affect the cell for a long
time. But the presence of 78 introns in this massive gene means there is a high risk of spontaneous and inherited mutations which could affect splicing, just because there are so many opportunities for this to happen. As one review rather pithily puts it: ‘The massive (2.4Mb) dystrophin gene, most of which is in its 78 introns, is a splicing accident waiting to happen, and it does so with an incidence of 1 in 3,000 live births.’
20
So, some cases of Duchenne muscular dystrophy are caused by splicing defects. However, in a large number of cases the disease is caused because critical regions of the gene, and hence the protein, are missing. But in recent years there has been a glimmer of hope for developing treatments for this invariably fatal disease. Perhaps counterintuitively, this has been based on developing drugs which
encourage
abnormal splicing in the dystrophin gene in affected boys.
The dystrophin protein acts like a kind of shock absorber in muscle cells. We can think of the dystrophin molecules as being like the springs in a mattress. In order for the mattress to remain supportive, the springs need to attach to the top and bottom of the mattress. If there has been a manufacturing fault, and the springs have been produced without the last ten centimetres, they won’t be able to attach to the top of the mattress. The more often you use the mattress, the less supportive and more distorted it will become.
It is quite common that Duchenne muscular dystrophy is caused by loss of internal regions of the dystrophin gene. When the gene is copied into RNA, the remaining regions are spliced together. Compared with the normal dystrophin gene, the mutant gene is now lacking some amino acids in the interior of the protein. But that’s not what really causes the biggest problem, as shown in Figure 17.6.
The amino acid code is read in blocks of three bases, as we have already seen. When the correct amino acid-coding regions (known as exons) join together, as in the normal gene, they produce a long messenger RNA molecule that codes for lots of amino acids. But
if wrong exons join together, they may be out of sync with each other, so that the blocks of three don’t run properly. A really simple example would be the following:
YOU MAY NOT SEE THE END BUT TRY
If we lose one letter, we rapidly start to lose the sense:
YOU MAY OTS EET HEE NDB UTT RY
This is called a frame shift. In messenger RNA the first impact is that the wrong amino acids are inserted into the growing protein chain. But quite soon, something even more dramatic happens. There will be a combination of three letters that act as a stop signal. At that point the ribosomes will cease adding amino acids, and the mutated protein is truncated.
This is what happens in the patients with deletions of certain regions of the dystrophin gene. In Figure 17.6, the frame of reading the three-base combination is indicated by the numbers under the boxes. As long as the number at the end of one box and the beginning of the next add up to three, the ribosome can keep reading the messenger RNA. But where the most common deletion occurs, this introduces a frame shift, which rapidly leads to a stop signal and a severely shortened protein chain.
One way around this would be to encourage the cell to skip one of the amino acid-coding regions after the deletion, because this would restore everything to the right reading frame. The end result would be a protein that has a bit missing internally, but can still function reasonably well. This might slow down progression of symptoms. This is shown using our bed spring analogy, in Figure 17.7. The dystrophin molecule will still be able to connect to the necessary proteins at either end. It won’t be quite as good
at absorbing shocks as the full-length protein. But it will be a lot better than one that can’t tether to the necessary cellular structures.

Figure 17.6
Representation of a key region where a mutation in the dystrophin gene can lead to a severely shortened protein molecule because of a shift in amino acid reading patterns when amino acid-coding regions 48 to 50 are lost from the DNA. In order to maintain the reading pattern, the numbers under each boundary must add up to three. If region 51 could be skipped in the mutant gene, the reading sequence would be restored. For simplicity, all the amino acid-coding regions have been drawn as the same size, although in reality they vary from one another.
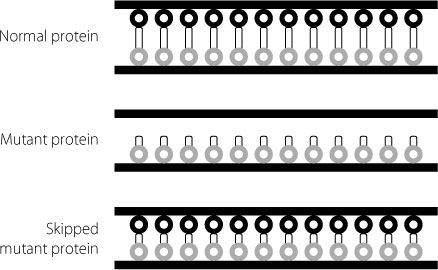
Figure 17.7
Schematic showing how a mutant dystrophin protein is unable to attach to the two sides of the cell membrane. The skipped version of the mutant protein, which is missing some internal sequences, can attach to the two sides of the membrane. Because it is shorter it isn’t as good a shock absorber as the normal protein, but it is much better than the original mutant.
The supporting evidence for this hypothesis looked good, and biotech companies began programmes to try to find a way of exploiting this knowledge. A company called Prosensa developed a drug that helped muscle cells to skip over amino acid-coding region 51 and eventually licensed this experimental drug to the pharmaceutical giant GlaxoSmithKline. In April 2013, GlaxoSmithKline published the results of a small trial in boys with the relevant form of Duchenne muscular dystrophy. Fifty-three boys were randomly assigned to two groups. One group received the drug, the other went through exactly the same procedures, but without actually receiving any of the experimental drug. This is known as a placebo procedure and is an important way of controlling for effects in clinical trials that are due not to the drug but to other effects such as increased optimism or patients getting better independently of
the drug. The boys were tested 24 and 48 weeks later. The test measured how far they could walk in six minutes.
After 24 weeks, the boys who received placebo had got worse, as we would expect for this disease. They couldn’t walk as far as when they entered the trial. But the boys who received the drug could walk more than 30 metres further than when the trial started. The boys were tested again after 48 weeks. The placebo group had deteriorated even more. In the six-minute walking test, their performance was almost 25 metres less than when the trial started. The boys who had been treated could walk over eleven metres further than when the programme began.
21