Pediatric Examination and Board Review (241 page)
Read Pediatric Examination and Board Review Online
Authors: Robert Daum,Jason Canel

(A) lead
(B) mercury
(C) thallium
(D) arsenic
(E) none of the above
13.
A 15-year-old girl comes to your clinic with the chief complaint of right facial weakness. The patient is unable to close her right eye. She was healthy until a couple of weeks ago when she developed an upper respiratory tract infection. She is otherwise fine. On physical examination, there is no other sign of weakness. She does not demonstrate fatigue on sustained motor testing. Her deep tendon reflexes are normal. A head CT performed in the ED is reportedly normal. Which of the following is the most likely cause of her symptoms?
(A) Möbius syndrome
(B) GBS
(C) Bell palsy
(D) myasthenia gravis
(E) cerebellar tumor
14.
Which of the following cervical nerves are injured in an Erb palsy?
(A) C5, C6
(B) C6, C7
(C) C7, C8
(D) C8, T1
(E) C5, C6, C7
15.
Which of the following cervical nerves are injured in a Klumpke paralysis?
(A) C5, C6
(B) C6, C7
(C) C7, C8
(D) C8, T1
(E) C5, C6, C7
16.
Which of the following are signs and symptoms of acute lead poisoning?
(A) seizures
(B) cerebral edema
(C) drowsiness
(D) irritability
(E) all of the above
17.
Which of the following vitamins can result in a sensory neuropathy if taken in large doses?
(A) vitamin A
(B) pyridoxine
(C) cobalamin
(D) vitamin C
(E) vitamin E
18.
Friedreich ataxia is transmitted by which mode of inheritance?
(A) as an autosomal recessive trait
(B) as an autosomal dominant trait
(C) by an X-linked mode of inheritance
(D) the disorder is the result of mutations of mitochondrial DNA
(E) not much is known about the genetics of Friedreich ataxia
ANSWERS
1.
(E)
This patient most likely has GBS, the diagnosis of which can be made by the history and physical examination. However, a number of confirmatory studies can be performed to support the diagnosis. These include MRI of the spine, lumbar puncture, and electrophysiologic studies, such as nerve conduction velocities and EMG. Enhancement of the cauda equina and lumbar roots following contrast administration is sometimes observed in GBS, indicating that an inflammatory process is indeed involved. An additional reason to perform an MRI of the spine with and without contrast is to exclude or confirm a transverse myelitis, which is in the differential diagnosis.
2.
(C)
The characteristic CSF finding in GBS is elevation of protein without significant pleocytosis (albuminocytologic dissociation). Approximately 10% of patients have 10-50 cells/mm
3
. Most of these cells are lymphocytes. A cell count greater than 50 cells/mm
3
should cause consideration of an alternative diagnosis. The protein level may be normal in the first few days but subsequently increases and peaks in 4-6 weeks. Opening pressure and glucose are usually normal.
3.
(B)
GBS or acute inflammatory demyelinating polyradiculoneuropathy is a disorder in which patients experience progressive motor weakness, areflexia, paresthesias, and elevated CSF protein typically following an upper respiratory tract infection. In 1990, Asbury and Cornblath proposed a set of diagnostic criteria. The criteria are:
Features required for diagnosis:
Progressive weakness of both arms and legs or more than 1 limb
Areflexia
Clinical features supportive of diagnosis:
Progression up to 4 weeks
Relative symmetry of signs
Cranial nerve involvement
Autonomic dysfunction
Recovery usually beginning 2-4 weeks after progression ceases
Absence of fever at onset
Laboratory features supportive of diagnosis:
Elevated CSF protein with less than 10 cells/μL
Electrodiagnostic features of conduction block or nerve conduction slowing
Autonomic dysfunction consists of sinus tachycardia, SVT, bradycardia, fluctuations in blood pressure, sphincter dysfunction, anhidrosis, and postural hypotension. Patients can experience respiratory difficulty requiring mechanical ventilation. Weakness usually starts in the lower extremities and ascends in a distal to proximal fashion. Cranial nerves can be affected with facial diplegia as the most common presentation. Paresthesias are a frequent early symptom and involve mild sensory loss and pain. Low back pain and myalgias are common. The pathogenesis of GBS involves immune-mediated segmental demyelination of peripheral nerves. By definition, the progression of symptoms ends by 4 weeks into the illness. If the progression lasts 4-10 weeks, then the term
subacute inflammatory demyelinating polyradiculoneuropathy
is used. If the progression is chronic or the patient experiences multiple relapses, the condition is termed
chronic inflammatory demyelinating polyradiculoneuropathy
.
4.
(C)
The Moro reflex can be present in children up to 5-6 months of age.
5.
(B)
The tonic neck reflex reaches a peak at approximately 2 months of age and should be absent by 6 months of age.
6.
(B)
The palmar grasp reflex disappears by 3-6 months of age. At this point, voluntary grasping should be evident. The plantar grasp reflex disappears much later by 15 months of age.
7.
(A)
Approximately 60% of children can mimic housework by 15 months of age with 100% of the normal children accomplishing this milestone by 17-18 months of age.
8.
(D)
Plasmapheresis and IVIG appear to be equally effective in the treatment of GBS or acute inflammatory demyelinating polyneuropathy. Corticosteroids are not effective. But they have been shown to be effective in the treatment of chronic inflammatory demyelinating polyneuropathy.
9.
(E)
The differential diagnosis of GBS is broad. The differential includes porphyria, transverse myelitis, acute myasthenia gravis, diphtheria, botulism, tick paralysis, hexane inhalation, Lyme disease, HIV infection, poliomyelitis, and hysteria. A good history and physical examination can help exclude some of these possibilities.
10.
(D)
GBS is regarded as a predominantly motor neuropathy with occasional sensory symptoms. Approximately three-fourths of the patients present with weakness. Pain and paresthesias are observed but to a lesser degree than the motor symptoms.
11.
(E)
Although plasmapheresis appears to be safe in children, there are a number of potential complications, including hypocalcemia, hemorrhage, hypotension, transfusion reactions, arrhythmias, and infection.
12.
(C)
Ingestion of certain heavy metals can result in a neurotoxic neuropathy. The heavy metals include lead, mercury, and thallium. Certain rat poisons and insecticides contain thallium. Ingestion may cause a sensory and motor peripheral neuropathy, nausea, vomiting, abdominal pain, and alopecia. Alopecia typically occurs 2-3 weeks after intoxication.
13.
(C)
This patient most likely has a Bell palsy or facial nerve paralysis. Bell palsy often follows an upper respiratory tract infection. Patients are unable to close the eyelid. Facial weakness develops over several hours to a few days. Lacrimation and taste may also be affected. Males and females are equally affected. Although approximately two-thirds of children recover fully without therapy, steroids have been used in the treatment of Bell palsy. Supportive eye care, including placement of an eye patch for protection and eye drops to prevent corneal dryness, is usually needed. The prognosis for recovery is good.
14.
(A)
Erb palsy is the most common brachial plexus injury in newborns. Damage to the upper trunk of the brachial plexus (C5 and C6 nerves) results in weakness of the upper arm. The patient presents with the humerus internally rotated and adducted. The elbow is extended and the wrist flexed.
15.
(D)
Klumpke palsy is a rare cause of brachial plexus injury in newborns. It results from damage to the lower brachial plexus (C8 and T1 nerves). Patients have weakness of the forearm extensors, wrist flexors, finger flexors, and intrinsic hand muscles. Horner syndrome can be present.
16.
(E)
Acute lead poisoning is now rare because the use of lead-based paints were banned in the United States in 1978. Acute lead encephalopathy initially presents with irritability and drowsiness followed by seizures and signs of increased intracranial pressure. Severe cerebral edema develops and can lead to irreversible neurologic injury.
17.
(B)
Large doses of pyridoxine can result in a sensory neuropathy with intact muscle strength. Paresthesias, autonomic dysfunction, diffuse sensory loss, and sensory ataxia can be observed.
18.
(A)
Friedreich ataxia is transmitted as an autosomal recessive trait. It is also one of the triplet repeat disorders. Patients with Friedreich ataxia develop a slowly progressive ataxia, cardiomyopathy, and sensory neuropathy. Deformities such as pes cavus and hammer toes are also observed (see
Figure 139-1
).
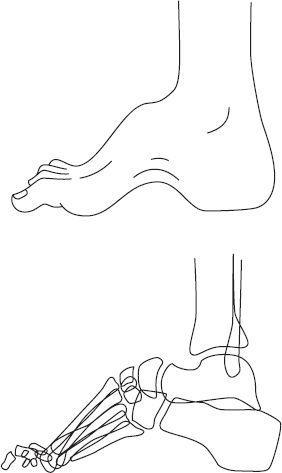
FIGURE 139-1
.
Cavus foot: clinical appearance and radiographic appearance. (Reproduced, with permission, from Skinner HB. Current Diagnosis & Treatment in Orthopedics, 4th ed. New York: McGraw-Hill; 2006: Fig. 11-19.)
 S
S
UGGESTED
R
EADING