Junk DNA: A Journey Through the Dark Matter of the Genome (29 page)
Read Junk DNA: A Journey Through the Dark Matter of the Genome Online
Authors: Nessa Carey

Not all the mutations that cause holoprosencephaly have been identified. Researchers studied the DNA from nearly 500 individuals who were affected by the condition. They found an unexpected change in a junk DNA region of one severely affected infant. This was a single base change, from C to T, in a region over 450,000 base pairs away from the morphogen gene.
6
The C to T change occurred in a block of ten base pairs that has been conserved since our ancestors diverged from the ancestors of frogs, over 350 million years ago. We can therefore surmise that this stretch of apparent junk has been maintained throughout evolution and has a function. In the case of this specific enhancer, the C binds a transcription factor protein.
c
Transcription factors are the proteins which are unusual because they recognise specific DNA sequences, usually in promoters, and bind to them. Binding of transcription factors to a promoter is essential for switching on a gene. The key transcription factor for this enhancer can bind to the ten-base-pair motif when the DNA contains a C in the appropriate position, but not when it contains a T.
This change from a C to a T in the enhancer wasn’t present in 450 unrelated healthy control individuals. That might make it seem very likely that this change was the cause of the problems in the patient, but it’s important to remember that it was also only seen once in about the same number of patients with the condition. The baby’s mother was unaffected, and as expected she had
the normal C base on both her chromosomes. But unexpectedly, the baby’s father had the same genetic sequence at the enhancer as his child. One chromosome had a C at the relevant position and the other had a T in the same place. But the father was completely unaffected by any symptoms of holoprosencephaly.
Although this might seem like strong evidence against a role for this C to T change, the situation isn’t that straightforward. In holoprosencephaly it’s quite common that there are lots of differences in a family, even where the mutation that causes the symptoms is in the morphogen gene itself. Up to 30 per cent of the family members with the mutation have no symptoms at all, and in others the symptoms may vary a lot from person to person. The first situation is known as variable penetrance, and the second is referred to as variable expressivity.
Unfortunately, these are classic cases where biologists identify a phenomenon, give it a fancy technical name and then stop thinking about it. These phrases are used to describe the phenomenon but we forget that we really don’t understand why it is happening. It’s a fascinating area that remains poorly understood. It’s possible that there are other subtle sequence variations in the genome that compensate for the effects of the DNA change in some people. This could include other enhancers working more strongly, and boosting expression of the morphogen. There may also be epigenetic compensation in some people, which nudges the expression of key genes in a certain direction. It may be a combination of both these factors, plus others that we have not yet identified.
But where we have this uncertainty – parent and child with the same genetic change but different symptoms – it’s vital to develop additional lines of evidence to support any hypothesis about the impact of the variant base. The researchers who identified the C to T change in the enhancer did exactly this, by testing the effect of this change in a mouse model. They showed that when the C was present, this stretch of junk DNA acted as an enhancer of
morphogen expression. But when the C was replaced by a T the region no longer acted as an enhancer, and levels of the morphogen never reached the critical levels in the brain.
Morphogens and the pancreas
The morphogen that is implicated in the development of extra digits or in the various forms of holoprosencephaly isn’t the only example of a human condition caused by a change in a regulatory region of DNA. There is a condition known as pancreatic agenesis, in which the pancreas fails to develop properly. Babies born with this condition often have severe diabetes.
7
This is because the pancreas is the organ that produces insulin, the hormone that allows us to regulate the levels of sugar in our blood.
The majority of families with pancreatic agenesis have a mutation in one particular transcription factor
d
,
8
but in a small number of affected families a different transcription factor is involved.
e
,
9
However, there are many cases where a child is born with unexplained pancreatic agenesis, when no one else in the family has ever been affected. Normally we might think that these cases have appeared randomly, perhaps as the consequence of something going wrong in development in response to an unidentified stimulus in the environment. But it became clear that most of these apparently sporadic cases occurred in families where the parents of the affected child were related to each other, typically cousins. This is known as consanguinity. When consanguinity is associated with higher rates of a disorder, we normally look for a genetic change. The change will be one where both copies of a chromosome carry the same variation. The reason why these conditions are more common in consanguineous couples is shown in Figure 15.1.
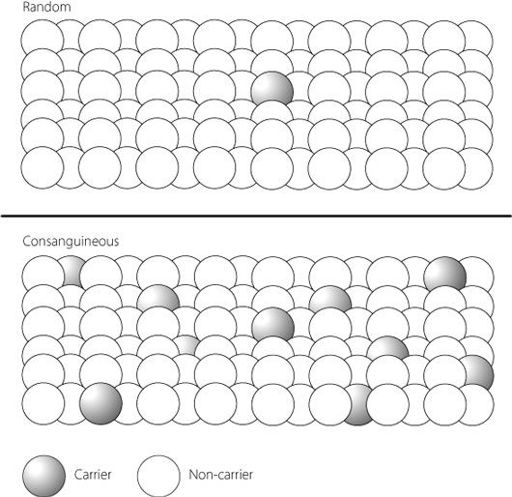
Figure 15.1
The upper part of the figure shows how a person who carries a rare genetic mutation is statistically relatively unlikely to meet another person with the same mutation in the general population. In their own family, however, it is much more likely that someone else will also have inherited the same mutation – a situation illustrated in the lower part of the figure. This is why rare recessive disorders (where the parents are asymptomatic carriers with one mutant gene each) present more commonly when the parents are related, for example first cousins.
Researchers isolated DNA from patients with the sporadic form of pancreatic agenesis and analysed all the protein-coding regions. They were unable to find any variations in sequence that could explain the disease. So they turned their attention to predicted Carrier Non-carrier
regulatory regions. There are, as we have seen, an awful lot of predicted regulatory regions in the human genome. In order to narrow down their search, the investigators studied what happens when stem cells differentiate into pancreas cells in culture. They looked for regulatory regions which carried epigenetic modifications normally associated with enhancer function, and which bound transcription factor proteins known to be important in the development of pancreas cells.
This narrowed the list of candidate regions to just over 6,000, a much more manageable number to analyse in depth. Four patients each had a change from an A to a G in a putative enhancer region of about 400 base pairs on chromosome 10. This region lay 25,000 base pairs away from one of the transcription factors that is known to be mutated in a small number of families with pancreatic agenesis. Seven out of ten unrelated patients all had the same change: the enhancer on both copies of chromosome 10 had a G where there is normally an A base. Two patients had other nearby mutations, and the tenth patient had lost the enhancer altogether. Nearly 400 unaffected people were analysed. None of them carried this A to G change.
The researchers showed experimentally that the region they had identified acted as an enhancer in developing pancreatic cells, and also showed that the region loses its enhancer activity when the A is changed to a G. In further experiments, they explored how this enhancer regulates its target gene. This is shown in Figure 15.2. Briefly, the enhancer loops out so that it lies close to its target gene. The enhancer normally binds transcription factors which help to switch on the target gene. But transcription factors only bind to certain DNA sequences. When the A is changed to a G the transcription factors can’t bind, and so they can’t switch on their target gene.
10
It’s a bit like going fishing. Drop a hook baited with a juicy worm into the lake, and a carnivorous fish will bite. Drop in a
hook baited with carrot and there won’t be so much as a nibble. Everything else is the same – the hook, the line, the sinker, the fish. But changing just one critical component (the bait) dramatically alters the chances of a successful catch.
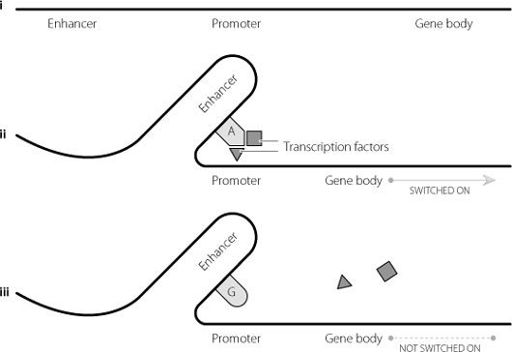
Figure 15.2
i shows the sequential positioning of an enhancer, promoter and gene body. In ii the DNA folds, bringing the enhancer close to the promoter. When the enhancer contains an A base in a specific position, the enhancer can bind specific proteins called transcription factors. These can activate the promoter and switch on the gene. In iii the A base in the enhancer is replaced by a G base, and the transcription factors can’t bind. This in turn means that they can’t activate the promoter, and the gene isn’t switched on.
Variations on a theme
It would be tempting to think that alterations in regions of junk DNA, which actually turn out to be regulatory regions, all have horrible consequences for the cell and the person. But that’s because it’s sometimes easier to look at abnormal situations than
normal ones. This is especially the case if we are assessing the difference between having a disease and being healthy. In the cases described above, the single base changes in the regulatory regions have had dramatic effects. But these types of variations are also responsible for situations which are much less binary, and are just a normal part of human diversity.
Consider pigmentation. Pigmentation is a complex trait, by which we mean it is influenced by lots of genes acting together. The end results in this case are eye, hair and skin colour. We all know by experience that humans vary enormously with respect to these features of our appearance. In addition to several genes contributing to pigmentation levels, there are also different variants of those genes, creating additional potential for variation.
11
One of the major variants is a single base difference, which occurs as either a C or a T. The T version is associated with higher levels of dark pigment, the C version with lower levels.
f
But this variation doesn’t lie in a protein-coding gene. It has been shown that the reason it affects pigmentation is because it is in an enhancer region, 21,000 base pairs away from the target gene. This target gene codes for a protein that is important for pigment production. We know this because mutations in this gene lead to a form of albinism where the affected individual can’t make pigment.
12
,
g
It has been shown experimentally that the enhancer loops to the target. Transcription factors that control the target bind with greater or lesser efficiency depending on the C or T base.
13
This is very similar to the situation outlined above for pancreatic agenesis, and uses pretty much the same mechanism as shown in Figure 15.2.
It’s quite likely that there are a lot of similar relationships between single base changes in junk DNA and the expression of protein-coding genes. This has implications for understanding
human diversity and human health and disease. There are a large number of conditions where we know genetics plays a role in whether or not a person develops a disorder. In these conditions, a person’s genetic background influences their likelihood of suffering from an illness, but doesn’t explain it entirely. The environment also plays a role; as, sometimes, does plain bad luck.
We can identify disorders with a genetic contribution by looking at how often a disease occurs in a family. Twins are particularly useful in this analysis. Let’s look at Huntington’s disease, a devastating neurological disorder caused by a mutation in one gene. If one twin has the condition, their identical twin will also always have the disease (unless they die early from an unrelated cause, such as a traffic accident). Huntington’s disease is 100 per cent due to genetics.