In Search of Memory: The Emergence of a New Science of Mind (42 page)
Read In Search of Memory: The Emergence of a New Science of Mind Online
Authors: Eric R. Kandel
Tags: #Psychology, #Cognitive Psychology & Cognition, #Cognitive Psychology

The circuit we traced is called a negative feedback circuit: a neuron excites an inhibitory interneuron that then inhibits the neuron that excited it in the first place. Could such an inhibitory feedback circuit be designed to hold an organism’s fear in check? To find out, we tested a genetically modified mouse whose receptors for gastrin-releasing peptide had been deleted, thus interrupting the inhibitory feedback circuit. We speculated that the resultant shift toward greater excitation might lead to increased, uncontrolled fear.
Consistent with our prediction, we found dramatically enhanced long-term potentiation in the lateral nucleus and a significantly enhanced and persistent memory of fear. The effect proved to be remarkably specific to learned fear: the same mutant mice showed normal innate fear on a variety of other tests. This finding is consistent with the fundamental distinction between learned and innate fear. Thus, a combined cellular and genetic approach allowed us to identify a neural circuit that is important for holding learned fear in check. The discovery could lead to the development of drugs that counteract learned fear associated with such psychiatric syndromes as post-traumatic stress disorders and phobias.
WHAT ABOUT THE OPPOSITE OF FEAR? WHAT ABOUT FEELING SAFE
, confident, and happy? In this context I cannot help being reminded of the first sentence of
Anna Karenina
, Leo Tolstoy’s novel about the tragic consequences of a socially unacceptable love affair: “Happy families are all alike; every unhappy family is unhappy in its own way.” Tolstoy here suggests, in a statement that has more literary than scientific power, that anxiety and depression can take many forms but that positive emotions—security, safety, and happiness—have common features.
With this idea in mind, Rogan and I explored the neurobiological characteristics of learned safety, presumably a form of happiness. We argued as follows. When a tone is paired with a shock, the animal learns that the tone predicts the shock. Thus if a tone and a shock are always given separately, the animal will learn that the tone never predicts the shock; instead, the tone predicts safety. When we carried out this experiment, we found exactly what we had predicted: when a mouse that was given shocks and tones separately heard the tone in a novel environment, it stopped acting defensively. It walked into the center of an open field as if it owned the place, showing no signs of fear (figure 25–5). When we looked in the lateral nucleus of mice that had undergone safety training, we found the opposite of long-term potentiation: namely, a long-term depression in the neural response to the tone, suggesting that the signal to the amygdala had been dramatically curtailed (figure 25–4).
We next asked whether safety training gives rise to a true sense of safety, an actual sense of self-confidence, or whether it simply lowers the baseline of fear that is always present in all of us. To distinguish between the two possibilities, we recorded from the striatum, an area of the brain normally involved in positive reinforcement and in feeling good. (This is the area activated by cocaine and other addictive drugs, which hijack the positive reinforcing neural system and entice a person to use the drug more often.) We found that neural activity in the striatum following a tone is not altered when the animal learns fear—that is, when it learns to associate the tone with a shock. But when an animal learns to associate the tone with safety, the response in the striatum is dramatically enhanced, consistent with the positive sensation of feeling safe.
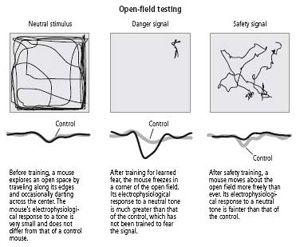
25–5
Effects of signals for learned fear and learned safety.
Our studies of learned safety have opened a new view of both positive feelings of happiness and security as well as negative feelings of anxiety and fear. They point to a second system deep in the brain that is concerned with positive emotions. Indeed, both the neurons in the thalamus that respond to the tone and the neurons in the lateral nucleus of the amygdala send connections to the striatum to convey information about contentment and safety. The striatum connects to many areas, including the prefrontal cortex, which inhibits the amygdala. So it is conceivable that by enhancing the signal in the striatum, learned safety not only enhances feelings of safety and security but also reduces fear by inhibiting the amygdala.
As these studies imply, we may be entering an era in which the molecular biology of cognition and emotion can open up ways of enhancing a person’s sense of security or self-worth. Might certain anxiety states, for example, represent a defect in the neural signals that normally convey a sense of security? Since the 1960s we have had medications that alleviate certain anxiety states, but these drugs are not useful for all anxiety disorders, and some of them, such as Librium and Valium, are addictive and therefore need to be monitored extremely carefully. Therapies that enhance the activity of the neural circuitry for safety and well-being might well provide a more effective approach to treating anxiety disorders.
C
an mouse models be used to investigate disorders that are even more complex, more serious and disabling than anxiety states? Can they be used to study schizophrenia, the most persistent and devastating mental disorder of humankind and the one most in need of new treatments?
Schizophrenia is, surprisingly, a fairly common disorder. It strikes about 1 percent of the population worldwide and seems to affect men slightly more frequently and more severely than women. An additional 2 to 3 percent of the general population has schizotypal personality disorder, often considered to be a milder form of the disease because patients do not manifest psychotic behavior.
Schizophrenia is characterized by three types of symptoms: positive, negative, and cognitive. The positive symptoms, which last at least six months, are odd or even bizarre behaviors and disturbances in mental functioning. They are most prominent during psychotic episodes, the phases of the illness in which patients are not able to interpret reality correctly. Patients then are unable to examine their beliefs and perceptions realistically or to compare them to what is actually occurring in the world around them. The hallmarks of this inability to interpret reality are delusions (aberrant beliefs that fly in the face of facts and that are not changed by evidence that the beliefs are unreasonable), hallucinations (perceptions occurring without an external stimulus, such as hearing voices commenting on one’s actions), and illogical thinking (loss of normal connections or associations between ideas, known as loosening of associations or derailment, which, when severe, results in incoherent thoughts and speech).
The negative symptoms of schizophrenia are an absence of certain normal social and interpersonal behaviors, accompanied by social withdrawal, poverty of speech, and a loss of the ability to feel and express emotions, called flattening of affect. The cognitive symptoms include poor attention and deficits in a form of explicit short-term memory known as working memory, which is critical for executive functions such as organizing one’s day, and planning and carrying out a sequence of events. The cognitive symptoms are chronic, persisting even during nonpsychotic periods, and are the most difficult aspects of the disease to manage.
Between psychotic episodes, patients exhibit primarily negative and cognitive symptoms: they behave eccentrically, are socially isolated, and have a low level of emotional arousal, impoverished social drive, poverty of speech, poor attention span, and lack of motivation.
Most people working on schizophrenia have recognized for some time that the entire spectrum of symptoms cannot possibly be modeled in mice. Positive symptoms cannot be modeled readily, because we do not know how to identify delusions or hallucinations in mice. It is equally difficult to model the negative symptoms. However, following the pioneering work of Patricia Goldman-Rakic in monkeys, carried out at Yale University, my colleagues Eleanor Simpson, Christoph Kellendonk, and Jonathan Polan wanted to know if it were possible to use mouse models to investigate the molecular basis of some aspects of the cognitive symptoms of schizophrenia. We thought we could model a key component of the cognitive symptoms—notably, the defect in working memory. Working memory has been well described and is known to be critically dependent on the prefrontal cortex, a part of the frontal lobe that mediates our most complex mental processes. We also believed that understanding the cognitive deficits would improve our understanding of how the prefrontal cortex functions during normal mental states.
STUDY OF THE PREFRONTAL CORTEX DATES TO
1848,
WHEN JOHN
Harlow described the now famous case of railroad foreman Phineas Gage. An accidental explosion drove a tamping iron through Gage’s prefrontal cortex. He survived the incident with his general intelligence, perception, and long-term memory intact, but his personality was changed. Before the accident, he was conscientious and hardworking; afterward, he drank a great deal and eventually became an unreliable drifter. Subsequent studies of people with injuries to the prefrontal cortex confirm that this region of the brain plays a critical role in judgment and long-term planning.
In the 1930s Carlyle Jacobsen, a psychologist at Yale, began to study the function of the prefrontal cortex in monkeys and provided the earliest evidence that it is involved in short-term memory. Four decades later, the British cognitive psychologist Alan Baddeley described a form of short-term memory that he called working memory because it integrates moment-to-moment perceptions over a relatively short period and relates those perceptions to established memories of past experiences, an essential feature in planning and executing complex behavior. Shortly thereafter, Joaquin Fuster at the University of California, Los Angeles and Goldman-Rakic linked Jacobsen’s work on the prefrontal cortex to Baddeley’s studies of working memory. They found that removing the prefrontal cortex of monkeys does not result in a generalized deficit in short-term memory but rather in a deficit in the functions that Baddeley described as working memory.
The finding that the prefrontal cortex is involved in the planning and execution of complex behaviors—functions that are disturbed in schizophrenia—led investigators to explore the prefrontal cortex of schizophrenic patients. Brain images revealed that metabolic activity in the prefrontal cortex is subnormal in these patients, even when they are not engaged in any specific mental activity. When normal individuals are challenged by a task that requires working memory, metabolic function in their prefrontal areas increases dramatically. The increase is much smaller in schizophrenic individuals.
Given that schizophrenia has a genetic component, it is perhaps not surprising that working memory is also moderately impaired in 40 to 50 percent of first-degree relatives (parents, children, and siblings) of patients with schizophrenia, even though these relatives lack clinical symptoms of the disease. Furthermore, the same relatives exhibit abnormal functioning of the prefrontal cortex, emphasizing the importance of this region in the genetic expression of schizophrenia.
The fact that the cognitive symptoms of schizophrenia resemble the behavioral defects seen when the frontal lobes are surgically disconnected from the rest of the brain in experimental animals caused us to ask: What are the molecular underpinnings of the defect in working memory in the prefrontal cortex?
MUCH OF WHAT WE KNOW ABOUT THE BIOLOGY OF SCHIZOPHRENIA
comes from the study of drugs that ameliorate the disorder. In the 1950s Henri Laborit, a French neurosurgeon, had the idea that the anxiety many patients experience before surgery might be caused by the body’s release of massive amounts of histamine. Histamine is a hormone-like substance produced in response to stress; it causes dilation of the blood vessels and a decrease in blood pressure. Laborit argued that the excess histamine might contribute to some of the undesirable side effects of anesthesia, such as agitation, shock, and sudden death. In his search for a drug that would block the action of histamine and calm patients, he came across chlorpromazine, which had just been developed by the French pharmaceutical firm Rhône-Poulence. Laborit was so impressed with the tranquilizing action of chlorpromazine that he began to wonder whether it might also calm agitated patients with psychiatric disorders. Two French psychiatrists, Jean Delay and Pierre Deniker, followed up on the idea and found that a high dosage of chlorpromazine indeed calmed agitated and aggressive patients with symptoms of schizophrenia.
In time, chlorpromazine and related drugs were found to be not only tranquilizers, calming patients without sedating them unduly, but also antipsychotic agents, dramatically reducing the psychotic symptoms of schizophrenia. These drugs, the first to be effective against a major mental disorder, revolutionized psychiatry. They also focused the interest of the psychiatric community on the question of how an antipsychotic agent produces its effects.
The first clue to chlorpromazine’s mechanism of action came from an analysis of one of its side effects, a syndrome resembling Parkinson’s disease. In 1960 Arvid Carlsson, a professor of pharmacology at the University of Göteborg in Sweden with whom I would later share the Nobel Prize, made three remarkable discoveries that provided critical insights into both Parkinson’s disease and schizophrenia. First, he discovered dopamine and showed it to be a neurotransmitter in the brain. Next, he found that when he lowered the concentration of dopamine in the brain of experimental animals by a critical amount, he produced a model of Parkinson’s disease. From this finding he argued that parkinsonism may result from a lowered concentration of dopamine in regions of the brain that are involved in motor control. He and others tested this idea and found that they could reverse the symptoms of Parkinson’s disease by giving patients additional dopamine.
In the course of these investigations, Carlsson noticed that when patients were given an overly large dose of dopamine, they developed psychotic symptoms resembling those seen in schizophrenia. This observation caused him to suggest that the underlying cause of schizophrenia is excessive dopamine transmission. Antipsychotic agents produce their therapeutic effect, he reasoned, by blocking dopamine receptors. This action reduces dopamine transmission along several critical neural pathways and thus lessens the consequences of excess dopamine production. Carlsson’s suggestion was later confirmed experimentally. Further support for his idea came from the finding that in treating patients, the antipsychotic drugs often produced Parkinsonian symptoms as a side effect of treatment, suggesting in still another way that these drugs block the action of dopamine in the brain.
In Carlsson’s view, the overactivity of dopamine-producing neurons was responsible for all of the symptoms of schizophrenia—positive, negative, and cognitive. He suggested that excess dopamine in the pathway to the hippocampus, to the amygdala, and to related structures might give rise to the positive symptoms, while excess dopamine in the pathway to the cortex, especially with that pathway’s abundant synaptic connections to the prefrontal cortex, might give rise to the negative and cognitive symptoms. In time, it became clear that all of the medications that alleviate the symptoms of schizophrenia target primarily a particular type of dopamine receptor, the D2 receptor. Solomon Snyder at Johns Hopkins and Philip Seeman at the University of Toronto both found a strong correlation between the effectiveness of antipsychotic drugs and their ability to block the D2 receptor. At the same time, however, it became clear that antipsychotic drugs help only with the positive symptoms of schizophrenia. They mitigate and even abolish delusions, hallucinations, and some types of disordered thinking without significantly affecting the negative or cognitive symptoms of the disease. This discrepancy was difficult to explain.
IN
2004
A NUMBER OF INVESTIGATORS DISCOVERED THAT ONE
genetic predisposition or susceptibility to schizophrenia is an abnormally large number of D2 receptors in the striatum, an area of the brain that, as we have seen, is usually involved in feeling good. Having an unusually large number of D2 receptors available to bind dopamine results in increased dopamine transmission. Simpson, Kellendonk, Polan, and I wanted to explore the role of this genetic susceptibility in producing the cognitive deficits of schizophrenia, so we engineered mice with a gene that expresses a superabundance of D2 receptors in the striatum. We found that such mice do indeed have deficits in working memory, consistent with Carlsson’s hypothesis.
We wanted to know why drugs that block D2 receptors fail to ameliorate the cognitive symptoms of schizophrenia, so we carried out another experiment, using genetic tools we had developed ten years earlier. Once a mouse reached adulthood, we turned off the transgene responsible for the production of excessive dopamine receptors and found that the defect in working memory was unabated. In other words, correcting the molecular defect in adult brains did not correct the cognitive deficit.
This result suggested that an overabundance of D2 receptors during development causes changes in the mouse brain that persist into adulthood. These changes might be the reason antipsychotic drugs do not work on the cognitive symptoms of schizophrenia. The overproduction of D2 receptors in the striatum exerts its impact early in development, well before the disease manifests itself, perhaps by producing fixed and irreversible changes in the dopamine system of some other part of the brain. Once this happens, the deficits in function of the prefrontal cortex, the structure involved in cognitive symptoms in the striatum, may no longer be reversible by reducing to normal the number of D2 receptors.
We have now tracked down at least one change that occurs in the prefrontal cortex as a result of the overproduction of D2 receptors: a decrease in the activation of another dopamine receptor, the D1 receptor. Earlier experiments by Goldman-Rakic had suggested that decreasing D1 receptor activation also decreases cyclic AMP, causing a deficiency in working memory.
These experiments demonstrate that genetically engineered mice may serve as valuable models in the study of complex psychiatric diseases by permitting us to break down the disease into simpler, more easily analyzed molecular components. Not only can we explore the genetic contributions to schizophrenia in mutant mice, we can also manipulate the environment of the mice, in utero and during early development, to examine what gene-environment interactions may trigger the onset of the disease.
DEPRESSION, ANOTHER COMMON ILLNESS THAT DESTROYS PSYCHIC
well-being, was first described in the fifth century
B.C.
by the Greek physician Hippocrates, who thought that moods depend on the balance of the four humors: blood, phlegm, yellow bile, and black bile. An excess of black bile was believed to cause depression. In fact, melancholia, the ancient Greek term for depression, means “black bile.” Although Hippocrates’ explanation of depression seems fanciful today, the underlying view that psychological disorders reflect physiological processes is becoming generally recognized.