Pediatric Examination and Board Review (142 page)
Read Pediatric Examination and Board Review Online
Authors: Robert Daum,Jason Canel

9.
(A)
The incidence of Down syndrome is approximately 1.0-1.2 in 1000 live births. The recurrence risk in chromosomally normal parents is 1% until the agerelated risk becomes higher than 1% (older than 40 years of age). Although the individual risk for Down syndrome is higher in older women, only 20% of all babies with Down syndrome are born to women older than the age of 35 because they account for only about 5% of all pregnancies. If the mother carries a balanced translocation, the recurrence risk is 10%; if the father is the carrier, the recurrence risk is 3-5%.
10.
(A)
See
Table 83-1
. The features described in answer B are most consistent with Down syndrome, those in C with trisomy 18, those in D with Prader-Willi syndrome, and those in E with Williams syndrome.
TABLE 83-1
Clinical Features of Common Autosomal Trisomies
| TRISOMY 21 DOWN SYNDROME | TRISOMY 18 EDWARD SYNDROME | TRISOMY 13 PATAU SYNDROME | |
Incidence | 1/800 | 1/8000 | 1/5000 |
Head shape | Microcephaly, brachycephaly, 3 fontanels | Microcephaly | Microcephaly, cutis aplasia, sloping forehead |
Eyes | Up-slanting palpebral fissures, epicanthal folds, Brushfield spots | Short palpebral fissures, corneal opacity | Micro-ophthalmia, hypotelorism, coloboma |
Ears | Small, low set | Low set, dysplastic | Low set, dysplastic |
Facial dysmorphic features | Protruding tongue, low flat nasal bridge | Small oral opening, micrognathia | Cleft lip and palate (60-80%) |
Extremities | Clinodactyly of 5th finger, simian crease, short metacarpals, wide gap between 1st and 2nd toe | Overlapping clenched fingers, hypoplastic nails, rocker bottom feet | Postaxial polydactyly hands and/or feet, hypoconvex nails |
Cardiac defect | 40% (AV canal most common) | 60% | 80% |
Kidney malformations | Polycystic kidney, horseshoe kidney | Polycystic kidney, duplicated ureters | |
Genitalia | Small penis, hypogonadism | Cryptorchidism, hypoplasia labia majora, prominent clitoris | Cryptorchidism, bicornuate uterus |
Neurological findings | Hypotonia, mild to moderate mental retardation | Feeble fetal activity, weak cry, postneonatal hypertonia, severe mental defect | Hypo- or hypertonia, holoprosencephaly, seizures, apnea, severe mental defect |
Dentition | Delayed eruption of teeth |
11.
(D)
See
Table 83-1
. Protruding tongue and large cheeks are seen in Down syndrome.
12.
(E)
Amniocentesis and CVS can both be used to obtain a prenatal karyotype to determine the presence of a trisomy or other chromosomal abnormality. CVS is done at 10-12 weeks of gestation and has a 1:100 risk of miscarriage; amniocentesis is done at 16 weeks of gestation and has a 1:200 risk of miscarriage. Maternal serum screening results of low levels of α-fetoprotein and unconjugated estriol, and elevated levels of human chorionic gonadotropin are associated with an increased risk of Down syndrome. Ultrasound can detect major fetal anomalies as early as 16 weeks of gestation, and those associated with increased risk for the presence of Down syndrome include increased nuchal thickening, congenital heart defects, duodenal atresia, and echogenic bowel.
13.
(D)
Turner syndrome has the karyotype 45,X and occurs in 1 in 8000 live births. Commonly associated features include short stature, webbed neck, low posterior hairline, shield chest, lymphedema of the extremities, cubitus valgus of elbow, and short fourth metacarpal and/or metatarsal. The physical phenotype is highly variable and often normal with the exception of short stature, which is typically not present at birth with a mean length of 146 cm (5th percentile). Approximately onethird of girls with Turner syndrome are diagnosed at birth because of lymphedema, one-third are diagnosed at ages 5-10 years because of short stature, and one-third are diagnosed secondary to delay or absence of puberty.
14.
(A)
The medical complications are highly variable with respect to severity and frequency and do not correlate with karyotype findings. Clinical manifestations include feeding problems in the newborn period, short stature, coarctation of the aorta, and/or bicuspid aortic valve (present in 17-45%), hypertension, mitral valve prolapse, hypothyroidism (occurring in 15-30% of adults), gonadal dysgenesis (90% require estrogen to initiate puberty and complete growth and estrogen and progesterone to maintain menses), ocular problems (strabismus), recurrent otitis media, and structural renal malformations in 40% (renal agenesis, horseshoe kidney, duplication of the collecting system, ureteropelvic and uretero vesicular obstruction). Cerebral findings include arteriovenous malformations but not agenesis of the corpus callosum or other structural brain malformations.
15.
(D)
Treatment with recombinant human growth hormone (GH) is effective in increasing height velocity. Many girls achieve heights of 150 cm or more with early initiation of treatment. Girls should be followed closely by an endocrinologist to track growth velocity and determine the optimal time to initiate GH. Children with Turner syndrome may have excessive weight gain, and careful monitoring of nutrition is essential to prevent obesity.
16.
(C)
Noonan syndrome is an autosomal dominant condition with 50% of patients having a mutation in the
PTPN11
gene on chromosome 12q24.1. Noonan syndrome occurs in boys and girls with equal frequency. It is often mistakenly referred to as male Turner syndrome because of the overlapping features. In both Noonan and Turner syndromes, patients have short stature, webbed neck, cardiac defects (coarctation of the aorta in Turner and pulmonary stenosis in Noonan), low posterior hairline, shield chest, wide-spaced nipples, and edema of the hands and feet (see
Figure 83-1
).
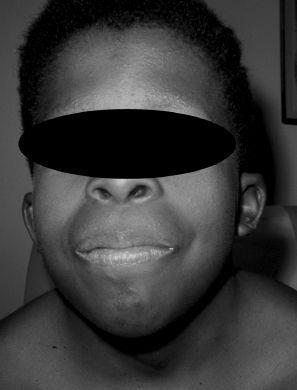
FIGURE 83-1.
Noonan syndrome: ptosis, hypertelorism, and low-set ears associated with valvular pulmonic stenosis. (Reproduced, with permission, from Fuster V, O'Rourke RA, Walsh RA, et al. Hurst’s the Heart. 12th ed. New York: McGraw-Hill; 2008: Fig. 12-17.)
17.
(C)
Klinefelter is caused by a sex chromosome abnormality, 47,XXY, and occurs with a frequency of 1:600 male live births. Common clinical features include tall stature that becomes evident by 5 years of age (newborns have normal growth parameters), eunuchoid habitus, slightly delayed motor and language milestones with IQ of 80-100, delayed sexual development, and infertility. The most frequently observed personality characteristics are shyness, nonassertiveness, immaturity, and a lack of confidence. Noonan syndrome has features that overlap with Turner syndrome and can occur in both boys and girls. Marfan syndrome is characterized by aortic root dilation, subluxed lens of the eye, and marfanoid body habitus. The 47,XYY pattern is characterized by tall stature, severe acne, and normal IQ with more aggressive behavior and behavioral problems. The testes are of normal size and function.
18.
(A)
Microdeletions such as DiGeorge syndrome are large deletions of chromosomal material but too small to be seen by routine karyotype. The resolution of karyotype is about 5-7 million base pairs (Mb) of genetic material. Microdeletions are often in the range of 3 Mb so are missed by routine karyotype and can only be detected by testing such as FISH. In the past, high-resolution karyotype was available as an independent test but is now considered standard for all karyotypes. Any karyotype would reliably pick up Down syndrome and would not need additional testing. Microarray technology now allows scanning the entire human genome for deletions and duplications as small as 100,000 bp. It would detect a deletion of the DiGeorge region, but in the case of a child with this specific heart defect, the likelihood of DiGeorge is high enough that focused testing with cheaper FISH technology is indicated. DiGeorge syndrome is characterized by heart defects, hypocalcemia, and immune function abnormalities. Approximately 40% of patients with TOF harbor this deletion. Williams syndrome is characterized by supravalvular aortic stenosis.
 S
S
UGGESTED
R
EADING
Cassidy SB, Allanson JE, eds.
Management of Genetic Syndromes.
3rd ed. New York, NY: Wiley-Liss; 2010.
Cohen WI, ed. Health Care Guidelines for Individuals with Down Syndrome: 1999 revision. Down Syndrome Research Foundation Web site.
http://www.denison.edu/collaborations/dsq/health99
. Accessed June 5, 2006.
CASE 84: A NEONATE WITH LETHARGY
A 7-month-old girl presents with lethargy, diaphoresis, pallor, and tachycardia. The mom states the child had a viral infection during the last few days with emesis one time, and she did not eat very well. The girl went to bed without eating dinner. This morning, the family had a difficult time arousing her and brought her straight to the office. She has no history of other health problems in the past. The family history is significant for two healthy siblings and one sibling who died at 4 months of sudden infant death syndrome (SIDS).
On examination, the vital signs show a respiratory rate of 60, heart rate of 160 bpm, and a blood pressure of 90/50 mm Hg. You note diaphoresis and pallor. The child is lethargic and difficult to arouse.