Resident Readiness General Surgery (78 page)
Read Resident Readiness General Surgery Online
Authors: Debra Klamen,Brian George,Alden Harken,Debra Darosa
Tags: #Medical, #Surgery, #General, #Test Preparation & Review

Figure 59-2.
Platelet adhesion and aggregation. GpIb receptors allow platelets to bind to the collagen in the subendothelium with the help of vWF. These are the receptors affected in Bernard-Soulier syndrome. GpIIa/IIIb receptors allow platelets to bind to one another with the help of fibrinogen and are decreased in Glanzmann thrombasthenia.
Coagulation factor deficiencies
Hemophilia A and hemophilia B resulting from factor VIII and factor IX deficiency, respectively, are considered to be the second and third most common hereditary bleeding diatheses. Both diseases are inherited on the X chromosome
and because they are coagulation factor abnormalities, patients will typically present with subcutaneous, intramuscular, or intra-articular hemorrhage following minor trauma. The degree of bleeding depends on the level of factor present in plasma. It is important to remember that factor VIII is the only coagulation factor that is not synthesized in the liver. Mild deficiencies in factor VIII can be treated with cryoprecipitate that contains high concentrations of the factor, or with desmopressin (DDAVP) that increases release of vWF and factor VIII from the endothelium. In most cases, however, deficiencies in factor VIII or IX are treated with the administration of factor concentrates. In general, factor levels need to be maintained at a minimum of 50% of normal in the intraoperative and postoperative periods until wound healing is complete in order to prevent complications related to hemorrhage. Other congenital factor deficiencies exist, but these are exceedingly rare and beyond the scope of this chapter.
3.
When a congenital coagulopathy is suspected, workup should proceed as follows:
A. Step 1—History and physical exam. Begin with a full history and physical exam focusing on any bleeding complications associated with previous operations, or a history of abnormal bleeding. It is important to remember that acquired causes of coagulopathy are more prevalent than congenital causes and the history and physical exam should be used to look for these diseases as well.
B. Step 2—Obtain a medication list. A full list of antiplatelet and antithrombotic medications taken by the patient should be obtained, and the patient should be asked about other medications that may interfere with platelet function such as aspirin, NSAIDs, fish oil, or H2 blockers.
C. Step 3—Screen for acquired causes of coagulopathy such as organ dysfunction. Liver disease may result in a coagulopathy as a result of poor synthetic function, and uremia secondary to renal failure results in abnormal platelet function by interfering with the action of both GpIIb/IIIa and GpIb receptors. The physical exam should not only look for evidence of abnormal hemostasis and hematopoiesis such as petechiae, hematomas, lymphadenopathy, and splenomegaly but also screen for stigmata of advanced liver and renal disease.
D. Step 4—If indicated, obtain appropriate laboratory studies. If there is ongoing concern for a bleeding diathesis, a complete blood count and prothrombin time (PT), international normalized ratio (INR), and PTT should be obtained. If there is concern for a platelet disorder despite a normal platelet count, a bleeding time assay and ristocetin cofactor assay should be obtained.
4.
In order to interpret laboratory data in the workup of congenital coagulopathies, one must understand which tests are representative of each part of the normal hemostasis pathway (see
Figure 59-2
).
Laboratory evaluation of platelet disorders
Platelet disorders can be evaluated with a complete blood count and bleeding time. If the complete blood count does not demonstrate thrombocytopenia and there is ongoing concern for a platelet disorder, a bleeding time assay can be obtained. To determine the bleeding time, a 9 mm long × 1 mm deep incision is made on the forearm and a blood pressure cuff is inflated to 40 mm Hg to create back pressure. Blood is blotted away every 30 seconds from the incision until bleeding stops. Although this test is extremely sensitive for picking up functional platelet disorders, it is quite difficult to standardize and is therefore not commonly used.
If there is suspicion for vWD, a ristocetin cofactor assay can be obtained. When added to normal blood, ristocetin will cause vWF to bind the GpIb receptor on platelets. In vWD, addition of ristocetin will not result in platelet clumping. It should be reiterated that vWD will result in a factor VIII deficiency and, therefore, some degree of PTT prolongation.
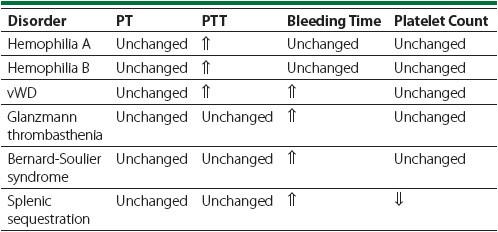
Table 59-2
shows the most common lab abnormalities associated with each of the above-mentioned bleeding diatheses.
Table 59-2.
Common Lab Abnormalities Associated With Bleeding Diatheses
Laboratory evaluation of factor deficiencies
PT: In general, the PT will be prolonged when there are low levels of factors in the extrinsic pathway and everything downstream of it, that is, VII, X, and
V, prothrombin, and fibrinogen. The INR is a standardized measurement of the PT and is used to monitor anticoagulation therapy.
PTT: The PTT is representative of the function of the intrinsic pathway and everything downstream of it, that is, factors VIII, IX, XI, XII, X, V, prothrombin, and fibrinogen. Therefore, hemophilia A and B are associated with a prolonged PTT.
Clearly, both the PT and PTT are affected by factors in the final common pathway and therefore any abnormality of the tenase complex and downstream of it will result in an elevation of both the PT and PTT.
A prolonged PT in the setting of a normal PTT ensures that the abnormality lies within the extrinsic pathway, upstream of the final common pathway. Only factor VII deficiency can cause this pattern of lab values.
Likewise, a prolonged PTT, but normal PT, implies that the abnormality lies within the intrinsic pathway, upstream of the final common pathway. This pattern will be seen with deficiency of factor VIII, IX, XI, or XII.
TIPS TO REMEMBER
The best screening test for a congenital coagulopathy is the preoperative history and physical exam.
Platelet disorders typically present with mucosal bleeding, epistaxis, prolonged bleeding after tooth extraction, and occasionally menorrhagia (in women).