Pediatric Examination and Board Review (248 page)
Read Pediatric Examination and Board Review Online
Authors: Robert Daum,Jason Canel

FIGURE 141-3.
Radiograph showing extensive calcinosis in the lower leg muscles and fascial planes of an adolescent with long-standing, poorly controlled dermatomyositis.
17.
(C)
HSP, the most common vasculitic syndrome of childhood, is characterized by the presence of a palpable purpuric rash (
Figure 141-4
). Distribution is primarily over the dependent areas of legs (especially prominent by the ankles) and buttocks, but it may also occur on the hands and arms. Ulceration of the lesions may occasionally occur. Recurrence of the purpuric rash, especially after increased physical activity, is common during the first 6 weeks. The rash is never associated with thrombocytopenia. Skin biopsy shows a leukocytoclastic vasculitis with IgA deposition. Subcutaneous edema may be present in younger children and occurs on the dorsum of hands and feet, periorbital areas, scalp, and scrotum. Arthritis, most commonly involving ankles and knees, occurs in 60-80% of children. Treatment of the arthritis is usually not necessary, although NSAIDs may be helpful. However, they should be used with caution because of the possible GI and renal involvement seen in HSP.
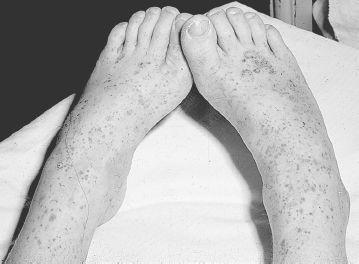
FIGURE 141-4
.
Palpable, purpuric lesions on the legs of a child with Henoch-Schönlein purpura. See color plates.
18.
(C)
GI involvement occurs in more than two-thirds of patients and can be mild or severe. It is usually characterized by colicky pain and may be associated with emesis and GI hemorrhage, either occult or gross. Abdominal pain is thought to be secondary to bowel wall edema. Mucosal ulceration may occur. When the GI symptoms are present before the rash (15-35% of cases), the diagnosis may be unclear and an acute surgical abdomen may be suspected. GI complications include intussusception (2-4%), and, rarely, bowel infarction, bowel wall perforation, pancreatitis, and hydrops of the gallbladder. Intussusception in HSP is usually ileoilial, as opposed to the more common ileocolic location seen in idiopathic intussusception. A barium enema, thus, will not often detect intussusception in HSP. A more helpful study to evaluate GI involvement in HSP is ultrasonography, which demonstrates bowel wall edema in children with GI symptoms, as well as complications such as intussusception and perforation. Although placebo-controlled studies are lacking, children with severe GI manifestations are often given CS. A reasonable treatment regimen is 1-2 mg/kg per day for 1 week, tapering off over the next few weeks. Intravenous CS administration may be required initially to assure systemic absorption.
19.
(D)
Renal involvement occurs in 20-40% of patients and generally presents within 3 months of the HSP diagnosis with microscopic hematuria. Mild proteinuria may also be present, but nephrotic syndrome, renal insufficiency, and hypertension are uncommon. UA should be performed weekly initially, and then at least monthly for the first 3 months after HSP diagnosis, and perhaps beyond in those with increased risk factors for renal disease: older age (>8 years of age); persistent rash; history of severe abdominal symptoms, initial abnormal urinalyses. Renal involvement progresses in 1-5% of patients and may result in renal failure. Early renal characteristics associated with persistent nephropathy or renal failure include proteinuria more than 1 g/day, nephrotic syndrome, and renal insufficiency. Renal biopsy findings range from mild mesangial proliferation in those with mild UA changes to severe crescentic glomerulonephritis in patients with more severe renal involvement. IgA deposition is present on immunofluorescence. Treatment of renal disease in HSP is controversial because of a lack of prospective multicenter trials. However, recent studies suggest improved outcomes of HSP renal disease with CS, alone or combined with an immunosuppressive medication such as cyclophosphamide, azathioprine, mycophenolate mofetil, or cyclosporine.
20.
(A)
KD is one of the most common vasculitides of childhood. Its pathology includes vasculitis and fibrinoid necrosis of medium-size arteries, especially the coronary arteries. The clinical characteristics for the KD classification criteria are well known and include the presence of persistent fever 39ºC or higher for 5 or more days with at least 4 of the following 5 features: nonspecific exanthema primarily on the trunk; oral changes (“strawberry” tongue and/or red, swollen, cracked lips); conjunctivitis (bilateral, bulbar, and nonsuppurative); cervical lymphadenopathy; extremity changes (edema and erythema of the hands and feet during the acute phase and/or desquamation of the palms and soles starting at the fingertips and toes during the subacute phase, approximately 2 weeks after clinical presentation). Some children, especially infants, may have incomplete or atypical KD, making the diagnosis more challenging and delaying treatment. Because children younger than 1 year of age are at increased risk for coronary aneurysms, any very young child with unexplained fever longer than 5 days associated with some features of KD should have an echocardiogram performed. In addition to the diagnostic features described, other common findings in KD include irritability (in part related to aseptic meningitis that occurs in 40% of patients), hydrops of the gallbladder, arthritis, and uveitis. Laboratory studies show markedly elevated acute phase reactants, white blood count and platelet counts, the presence of anemia, mild liver enzyme elevation, and negative cultures and ANA. Once the diagnosis of KD is made, or is highly suspected, IVIG, 2 g/kg, should be administered. This treatment is especially effective when given within 10 days of presentation, usually resulting in immediate defervescence and a marked decrease in the risk of developing coronary aneurysms (from >20% to <5%). If clinical symptoms persist, retreatment with IVIG is indicated. Some patients require further treatment, usually high-dose intravenous methylprednisolone. High-dose aspirin (80-100 mg/kg/day) is also given during the acute phase and then decreased to antiplatelet dosing (3-5 mg/kg) until acute-phase reactants and platelet levels return to normal. Low-dose aspirin is continued indefinitely in those with coronary aneurysms. An echocardiogram should be performed when the KD diagnosis is made and then repeated 6-8 weeks later. De novo aneurysms rarely develop more than 8 weeks after disease onset. Long-term sequelae occur most often in patients who were diagnosed with giant aneurysms (internal vessel diameter ≥8 mm) during the acute illness. These patients may develop vessel stenosis and occlusion, leading to ischemia and/or infarction. In industrialized countries, KD is the leading cause of acquired heart disease in children.
21.
(E)
Scleroderma is classified as (1) systemic disease that includes diffuse cutaneous systemic scleroderma (DCSS, or systemic sclerosis) and limited scleroderma (or CREST, which is very rare in pediatrics); (2) localized disease (see answer 22). The pathology of systemic and localized scleroderma is similar, with early inflammatory infiltrate in the skin and other involved tissues, followed by increased fibroblast and collagen deposition. A vasculopathy with endothelial cell injury is prominent in this disease. The etiology is unclear. DCSS is a very rare but potentially devastating illness in children. Raynaud, present in more than 90%, is often severe, resulting in digital ulcerations and gradual loss of distal tufts (
Figure 141-5
). Initially, tense swelling with subcutaneous edema, especially of the fingers and arms, is prominent. Then the skin gradually tightens down, resulting in marked limitation in finger and large joint range. Telangiectasias and subcutaneous calcification are common. Arthritis, especially of the small joints, and myositis can occur. Pulmonary disease is present in most patients and can be asymptomatic or characterized by dyspnea on exertion or dry cough. Lung involvement may be primarily parenchymal with fibrosis, vascular leading to pulmonary hypertension, or a combination of both. Serial pulmonary function tests are indicated during the course of the disease to evaluate for restrictive disease and decreased diffusion. DCSS can affect the entire GI tract, but the most prominent problems are lower esophageal dysfunction resulting in reflux, gut hypotonia, and malabsorption. Radiographic studies, especially to assess esophageal function, may be helpful in guiding treatment. Renal disease is primarily vascular and can lead to renal crisis with malignant hypertension. Cardiac involvement may be primary or may be secondary to pulmonary hypertension. Periodic echocardiograms should be performed. Laboratory studies are not helpful in making a diagnosis of DCSS or in assessing disease status. Most patients do have a high-titer positive ANA. Management of DCSS is challenging and often customized based on clinical involvement. Treatment generally includes intensive rehabilitation (especially occupational therapy), H2 blockers and proton pump inhibitors for reflux, calcium channel blockers for Raynaud, angiotensin-converting enzyme inhibitors for hypertension, and various anti-inflammatory and immunosuppressive medications (which are, unfortunately, frequently not very efficacious) to treat the fibrotic and vascular complications of this disease.
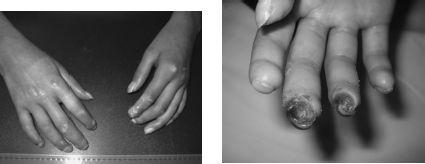
FIGURE 141-5.
(left) Diffuse cutaneous systemic scleroderma: taut, shiny and thickened skin on hands/fingers with small ulcerations and early loss of distal tufts. (right) Same patient 2 years later with large distal ulcers and further loss of distal tufts.
22.
(B)
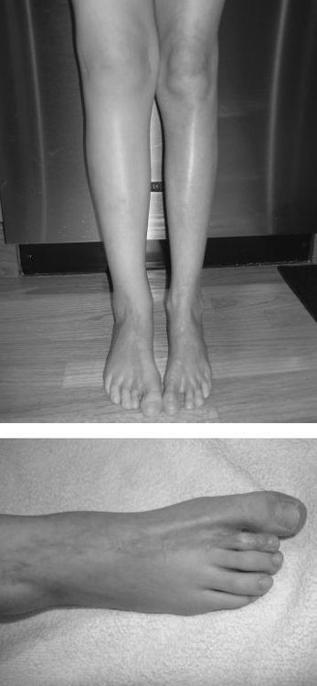
Localized scleroderma is more common than systemic sclerosis and limited scleroderma (or CREST), which is rare among children. Often referred to as morphea, this group of disorders is characterized by involvement of skin and subdermal tissues. There is absence of systemic complications and Raynaud. Several subtypes of morphea are described, varying from mild and scattered superficial plaques to more severe generalized and deep morphea. Linear scleroderma, the most common form of localized scleroderma/morphea, usually involves one limb, although 2 or more limbs may be involved. Lower extremities are affected more often than upper. Involvement extends through subcutaneous tissue and muscle to underlying bone, leading to inhibited growth of the affected limb, severe muscle atrophy, and flexion contractures (
Figure 141-6
). Linear scleroderma of the face, or en coup de sabre, may result in facial hemiatrophy, dental/orthodontic problems, and major cosmetic concerns. Parry-Romberg syndrome refers to progressive facial hemiatrophy without the classic “en coup de sabre” lesion, often complicated by seizures and trigeminal neuralgia. Laboratory studies in localized scleroderma are not diagnostic, although the ANA is often positive, as is singlestranded DNA. No treatment is indicated for superficial morphea, whose lesions often regress over a few years. Deep and generalized morphea, as well as linear scleroderma, often respond to treatment with systemic CS and methotrexate. Physical and/or occupational therapy is indicated in children with linear scleroderma.