Pediatric Examination and Board Review (149 page)
Read Pediatric Examination and Board Review Online
Authors: Robert Daum,Jason Canel

(C) methylprednisolone therapy
(D) intravenous spectrin replacement therapy
(E) monthly intravenous gammaglobulin infusions
18.
Postsplenectomy patients are not at increased risk for infections from which of the following bacteria?
(A)
Streptococcus pneumoniae
type 19A
(B)
Haemophilus influenzae
(C)
Mycoplasma pneumoniae
(D)
Escherichia coli
(E)
Streptococcus pneumoniae
type 3
ANSWERS
1.
(C)
Neonatal jaundice occurs in approximately twothirds of infants and is defined by bilirubin levels higher than 5 mg/dL. Bilirubin is generated as one of the products of the breakdown of hemoglobin, and the conjugation of bilirubin to bilirubin glucuronide occurs in the liver. Neonatal unconjugated hyperbilirubinemia is therefore the result of either increased bilirubin production or decreased conjugation. Neonatal hemolytic anemias, with increased bilirubin production, can result in severely abnormal unconjugated hyperbilirubinemia levels in neonates. Blood group mismatches because of ABO or Rh mismatches are common causes of neonatal hemolytic anemia and unconjugated hyperbilirubinemia. Other causes of unconjugated hyperbilirubinemia include hemolytic anemias because of red blood cell membrane or enzyme defects and increased red blood cell turnover associated with polycythemia, internal hemorrhages such as cephalohematomas or intraventricular hemorrhages. Decreased bilirubin conjugation as a result of Crigler-Najjar or Gilbert syndromes also result in unconjugated hyperbilirubinemia. Biliary atresia, α
1
-antitrypsin deficiency, and Caroli syndrome are all causes of neonatal conjugated hyperbilirubinemia.
2.
(C)
Evaluation for suspected neonatal hemolytic anemias should include evaluation of the complete blood count along with indirect and direct Coombs tests and maternal and neonatal blood types. In the absence of excessive bleeding or bruising, there is no indication for coagulation studies. The complete blood count will reveal the degree of anemia, if any, and the peripheral smear will demonstrate the morphologic features of the red blood cells that could suggest underlying etiologies. The Coombs tests will evaluate whether or not there are antibodies that could be contributing to autoimmune or alloimmune hemolysis. Neonatal and maternal blood types are needed to evaluate the possible presence of ABO and Rh incompatibility.
3.
(B)
Mothers with Rh negative blood type develop antibodies against the Rh antigen after exposure during pregnancy to an Rh-positive fetus or after transfusion with Rh-positive blood. In the presence of an Rh-positive fetus, the mother’s antibodies can cross the placenta and destroy fetal red blood cells, resulting in neonatal hemolytic anemia. Rh hemolytic disease can be prevented with high titer Rho(D) immune globulin treatment for Rh-negative mothers who have been exposed to Rh-positive infants. Similarly, mothers with type O blood have antibodies against antigens for blood types A and B that can react to and destroy fetal red blood cells with these blood type antigens. However, a fetus that also possesses type O blood will not be susceptible to hemolysis from these antibodies. A mother with type AB blood has no antibodies to blood group antigens, and therefore the fetus is not exposed to any anti-red blood cell antigen antibodies. Despite the presence of these antibodies, only 33% of infants with ABO “mismatch” will have a positive direct Coombs test, and of those, only 20% will develop jaundice from excessive hemolysis.
4.
(C)
Crigler-Najjar syndrome is caused by the absence of glucuronyl transferase and results in severe indirect hyperbilirubinemia. Pyruvate kinase and G6PD are both red blood cell enzymes that, when deficient, can result in neonatal hemolytic anemia. Hereditary spherocytosis, as well as other syndromes with red blood cell structural abnormalities, such as hereditary elliptocytosis and paroxysmal nocturnal hemoglobinuria, can also result in neonatal hemolytic anemia. A maternal-fetal blood type mismatch such as ABO incompatibility results in alloimmune hemolytic anemia, with the mother’s antibodies reacting to and destroying the neonatal red blood cells.
5.
(C)
Erythroblastosis fetalis, or hydrops fetalis secondary to Rh hemolytic disease, is characterized by severe fetal hemolytic anemia as a result of anti-Rh antibody from an Rh-negative mother crossing the placenta and attacking fetal Rh-positive red blood cells. The hemolysis results in intrauterine hyperbilirubinemia, which can be detected in amniotic fluid samples. Furthermore, the severe anemia results in high output cardiac failure with anasarca and peripheral edema. Hepatosplenomegaly as a result of extramedullary hematopoiesis also occurs. Nucleated red blood cells, or “erythroblasts,” are elevated as a result of fetal marrow hyperproduction to compensate for the anemia (see
Figure 87-1
).
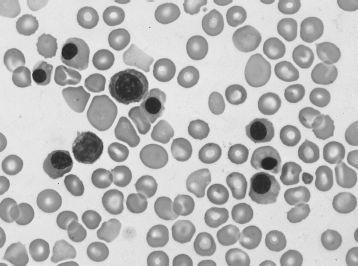
FIGURE 87-1.
Hemolytic disease of the newborn (erythroblastosis fetalis). Polychromatophilic cells, spherocytes, and circulating erythroblasts. (Reproduced, with permission, from Lichtman MA, Beutler E, Kipps TJ, et al. Williams Hematology, 7th ed. New York: McGraw-Hill; 2006: Plate II-2.)
6.
(B)
Red blood cell transfusions or exchange transfusions are often required for infants suffering from Rh-hemolytic disease but must be cross-matched against the mother and not the infant because the mother’s antibodies are the source of the hemolytic anemia. Fresh-frozen plasma (FFP) has no role in the management of the hemolytic anemia in the infant. Treatment of the mother with Rho(D) immune globulin before delivery can reduce the autoimmune hemolysis, but intravenous immunoglobulin does not have the same beneficial effect.
7.
(C)
The direct Coombs test takes the patient’s red blood cells and places them in the presence of complement proteins in vitro. The occurrence of hemolysis confirms the presence of antibodies directly bound to the patient’s red blood cells. The indirect Coombs test evaluates the patient for antibodies to red blood cells that are circulating freely in the serum. The Coombs tests do not evaluate antibodies against any other types of blood cells.
8.
(C)
Infants in their first few days of life cease producing new red blood cells as a result of the oxygen-rich environment (relative to in utero) and decreased responsiveness of their bone marrow to erythropoietin. The hemoglobin levels fall to a nadir of 9-10 g/dL at 10-12 weeks of age, at which time erythropoiesis resumes. Premature infants or infants with other causes of neonatal anemia have an earlier onset of physiologic anemia with a lower nadir level of hemoglobin, often down to 6-8 g/dL at 4-8 weeks of age.
9.
(B)
Careful observation is generally all that is required for physiologic anemia because the infant’s erythropoietic system matures and the anemia resolves. Iron and folic acid supplementation are not indicated for isolated physiologic anemia, and transfusions are only indicated for severe symptomatic anemia, which does not occur with isolated physiologic anemia. Blood transfusions can suppress the endogenous erythropoietin production and delay recovery from physiologic anemia.
10.
(B)
The presence of G6PD deficiency is most common in African populations and populations that dwell or dwelled around the Mediterranean Sea, with increased frequency also in southern Asian populations and American Indians. G6PD deficiency occurs in approximately 12% of African American males; in Southeast Asia the incidence is approximately 6%. Northern Europeans have the lowest incidence of G6PD deficiency among the populations listed.
11.
(A)
G6PD deficiency is an X-linked disorder. Affected males have a single mutated copy of the G6PD gene; affected females are usually compound heterozygotes with 2 different mutant G6PD gene alleles. Deficiency of G6PD in African Americans is most commonly the result of a mutation that renders the enzyme unstable, leaving new red blood cells with relatively normal enzyme levels but old red blood cells with nearly absent enzyme levels and with increased susceptibility to oxidant stress. The mutations in the G6PD gene that occur in other populations, particularly the Mediterranean and Middle Eastern populations, generally result in absent G6PD expression. These patients are more susceptible to oxidant-induced red blood cell lysis and are also susceptible to hemolysis induced by fava beans (termed
favism
).
12.
(A)
Heinz bodies (see
Figure 87-2
) are intracellular inclusions of oxidized and degenerated hemoglobin seen in red blood cell enzyme deficiencies such as G6PD deficiency. The inclusions are often attached to the red blood cell membrane and can be “eaten” by splenic macrophages, resulting in the characteristic “bite cells” of red blood cell enzyme deficiencies. Autoimmune hemolytic anemia is characterized by microspherocytosis, but there are no intracellular inclusions. Neonatal alloimmune thrombocytopenia has no unusual red blood cell features. Sickle cell disease has sickled red blood cells and often has Howell-Jolly bodies from splenic hypofunction, but it is not associated with Heinz bodies.
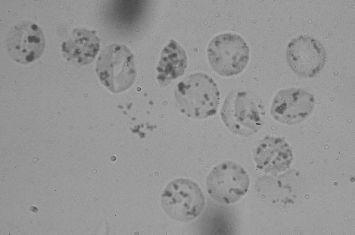
FIGURE 87-2.
Heinz bodies. Blood mixed with hypotonic solution of crystal violet. Precipitates of denatured hemoglobin within the cells. (Reproduced, with permission, from Lichtman MA, Beutler E, Kipps TJ, et al. Williams Hematology, 7th ed. New York: McGraw-Hill; 2006: Plate IV-4.)
13.
(D)
Hemolysis in patients with G6PD deficiency is stimulated by oxidative stress, which can be caused by stress from infections, diabetic ketoacidosis, or by medications and chemicals such as primaquine, sulfa drugs, and methylene blue. Other chemicals that can trigger hemolysis include dapsone, nitrofurantoin, trinitrotoluene, naphthalene, and acetanilid. Fava beans can stimulate hemolysis in certain populations with specific forms of G6PD mutations, such as those that occur in Mediterranean populations. Acetaminophen is not associated with hemolytic crises in patients with G6PD deficiency.
14.
(C)
Hemolytic crises in patients with G6PD deficiency and other hemolytic anemias are characterized by fatigue, pallor, scleral icterus and jaundice, splenomegaly, and hemoglobinuria. Laboratory findings include anemia with increased free plasma hemoglobin and decreased plasma haptoglobin. Pulmonary infiltrates are not associated with hemolytic crises.
15.
(B)
Hereditary spherocytosis is the most common structural red blood cell defect and affects approximately 1 in 5000 people. The most common cause of hereditary spherocytosis is a defect in spectrin, a protein responsible for the structural integrity of the red blood cell membrane. Defects in associated proteins, such as band 3, protein 4.2, and ankyrin, can also play a role in the defect but act via a relative spectrin deficiency. Hereditary spherocytosis is most common in northern Europeans and is characterized by anemia, jaundice, and splenomegaly. Most cases are inherited in an autosomal dominant fashion, but approximately 10% are inherited as a recessive trait. Hemoglobin deficiencies do not cause spherocytosis, and pyruvate kinase is a red blood cell enzyme that is unrelated to the structural features of the red cell membrane.